 |
A nine-year-old female presented to the clinic for evaluation of poor vision in both eyes. Her grandmother said the girl’s vision had always been poor. At a visit four years earlier, her acuity was measured at 20/70.
The grandmother felt it was now significantly worse and revealed that the patient was born at 37 weeks, weighed 6.8 pounds and had a normal birth and development. However, when speaking directly to the girl, she demonstrated poor mentation and was unable to recall simple things (i.e., the date of her birthday).
Diagnostic Data
Entrance testing revealed a visual acuity of light perception (LP) in both eyes. Her pupils were equally round and reactive to light. There was no afferent pupillary defect in either eye. Confrontation visual fields were unobtainable.
Extraocular movements were full, but there was nystagmus in both eyes. Intraocular pressures (IOP) were within normal limits, measuring 15mm Hg OD and 16mm Hg OS.
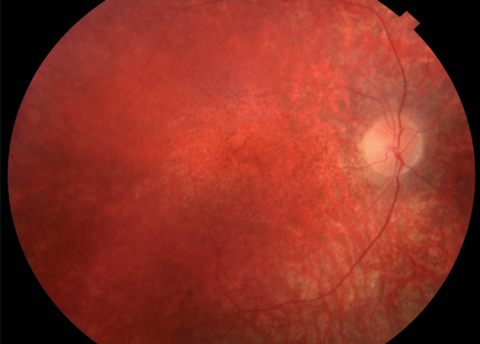
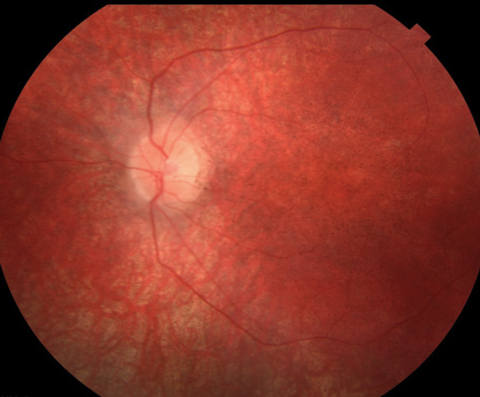
Anterior segment was within normal limits and unremarkable in both eyes. Dilated fundus examination revealed a normal vitreous OU. Other changes can be seen in the fundus photos (Figure 1). Fundus autofluorescence (FAF) and optical coherence tomography (OCT) were also obtained (Figure 2).
 |
 |
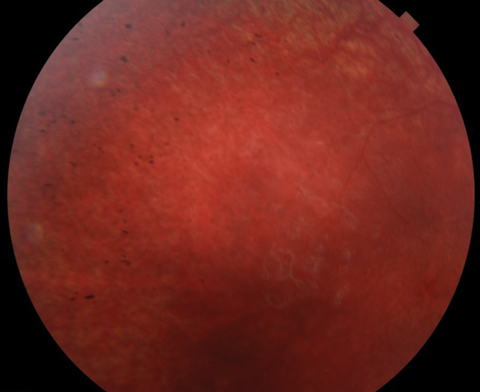 |
| Fig. 1. At top, this fundus photo shows our nine-year-old patient’s right eye. The middle image shows her left eye, while the bottom shot shows the periphery of the right eye. The same appearance was noted in the periphery of the left eye. |
Take the Quiz
1. What is the likely diagnosis?
a. Leber’s congenital amaurosis.
b. Retinitis pigmentosa.
c. Stargardt’s macular dystrophy.
d. Batten disease.
2. Which condition below is associated with an accumulation of lipofuscin in neuronal tissue, including the brain, retina and peripheral nerves?
a. Neuronal ceroid lipofucinosis.
b. Retinitis pigmentosa.
c. Multiple sclerosis.
d. Age-related macular degeneration.
3. In regards to FAF imaging,
hyperautofluorescence is considered indicative of______.
a. Cystoid changes.
b. Loss of lipofuscin.
c. Increased lipofuscin or storage material such as lipoproteins and hydrophobic peptides.
d. None of the above.
4. What is the likely prognosis for this condition?
a. Continued profound vision loss to no light perception.
b. Slow improvement of visual acuity.
c. Geographic atrophy of the retina and retinal pigment epithelium.
d. Stabilization of acuity to 20/400.
Discussion
Based on the profound vision loss and retinal changes, coupled with evident developmental problems, our patient likely has the neurodegenerative condition Batten disease.1 Specifically, the condition is classified as a neuronal ceroid lipofucinosis (NCL) and represents a group of disorders that have a characteristic accumulation of liposfuscin in the neuronal tissues including the brain, retina and peripheral nerves.2 The clinical ocular features of Batten disease include optic nerve pallor, central macular mottling, attenuated arterioles and peripheral retinal granularity.3 All these retinal changes were noted on clinical examination of our patient.
The literature describes the following four forms, often collectively referred to as Batten disease:1
• Haltia-Santavouri disease has an infantile onset.
• Jansky-Bielschowsky disease has an infantile onset.
• Batten-Mayou Syndrome has a juvenile onset.
• Kufs disease has an adult onset.
These children suffer from severe psychomotor deterioration that leads to vegetative states, seizures, profound visual loss secondary to retinal degeneration and, ultimately, premature death.1 NCL disorders are inherited in an autosomal recessive manner.2 The rate of progressive vision loss that occurs in NCL disorders is extremely rapid, with profound vision loss occurring in a matter of months.1
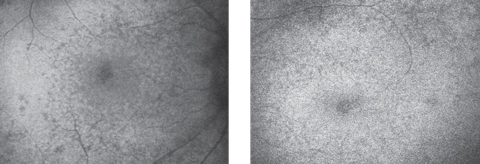 |
| Fig. 2. Autofluorescence of the right (at left) and left maculae. Click image to enlarge. |
Identification
The differential diagnosis for this patient includes retinitis pigmentosa (RP) and Stargardt’s disease (SD). RP represents a group of retinal degenerative diseases that begin by affecting the rod photoreceptors and eventually lead to degeneration of the cone photoreceptors.4 SD is the result of a mutation in the ABCA4 gene that leads to accumulation of byproducts within the retinal pigment epithelium (RPE) that cause dysfunction and death of the photoreceptors.5
Appropriate ancillary testing for any patient with a suspected hereditary retinal disease includes full-field electroretinography (ERG), electrooculography (EOG), dark adaptation, perimetry testing, OCT, FAF and genetic testing. ERG studies of Batten disease patients are often abnormal early on and eventually becomes flat or undetectable. FAF shows an accumulation of storage material that hyperautofluoresces. This storage material appears similar to ceroid and liposfuscin, but is actually a mixture of lipoproteins and hydrophobic peptides.1,2 Hypoautofluorescence on autofluorescence imaging is indicative of atrophy.6 Lastly, OCT imaging will show small hyper-reflective bodies at the RPE level.1,2
In the past, biopsies from the brain and full-thickness rectal biopsy were the standard protocol for diagnosing NCLs. These biopsies looked for inclusion bodies in the cells of these tissues. Currently brain biopsies are no longer justifiable for the diagnosis of NCL. Rather muscle biopsy may represent the best approach to ultrastructural diagnosis of the disease; however, muscle biopsies are not ideal for differentiating between the four forms of NCLs as the fibers only show curvilinear inclusions.1
As patients with Batten disease may be nonresponsive and have potentially poor prognosis, genetic testing is necessary to confirm the diagnosis. Our patient was referred to a genetics specialist the same day. The genetics specialist counseled the patient and family on the possible results. It was also recommended that siblings of the patient be genetically tested as well.
Until recently, there has not been a treatment for Batten disease; however, on April 27, 2017, the FDA approved the drug Brineura (cerliponase alfa, BioMarin) as a treatment option for the specific form of Batten disease known as Jansky-Bielschowsky disease. It is the first FDA-approved treatment shown to retard ambulatory loss in pediatric patients ages three and older suffering from the late infantile NCL type 2. Brineura is an enzyme replacement therapy.7 Its main component, cerliponase alfa, is the recombinant form of human TPP1, the specific enzyme that is deficient in those suffering from this form of Batten disease.7
Dr. Diego is a resident at Bascom Palmer in Miami.
1. Ryan S, Schachat A, Wilkinson C. 5th ed. Retina. London: Saunders/Elsevier;2013. |
