 |
History
A 16-year-old African-American female presented for a routine eye exam. She had no ocular or systemic history and denied taking medication of any kind. She had no known allergies. She was a foster child, so her family history was unobtainable.
Diagnostic Data
Her entering best-corrected visual acuities were 20/20 OU at distance and near. Her external examination was normal. No evidence of afferent pupil defect was present. Biomicroscopic examination of the anterior segment demonstrated normal tissues with Goldmann applanation pressures measuring 16mm Hg OU. However, her dilated fundus findings did reveal an abnormality.
Your Diagnosis
Does the case presented require any additional tests, history or information? What steps would you take to manage this patient? Based on the information provided, what would be your diagnosis? What is the patient’s most likely prognosis?
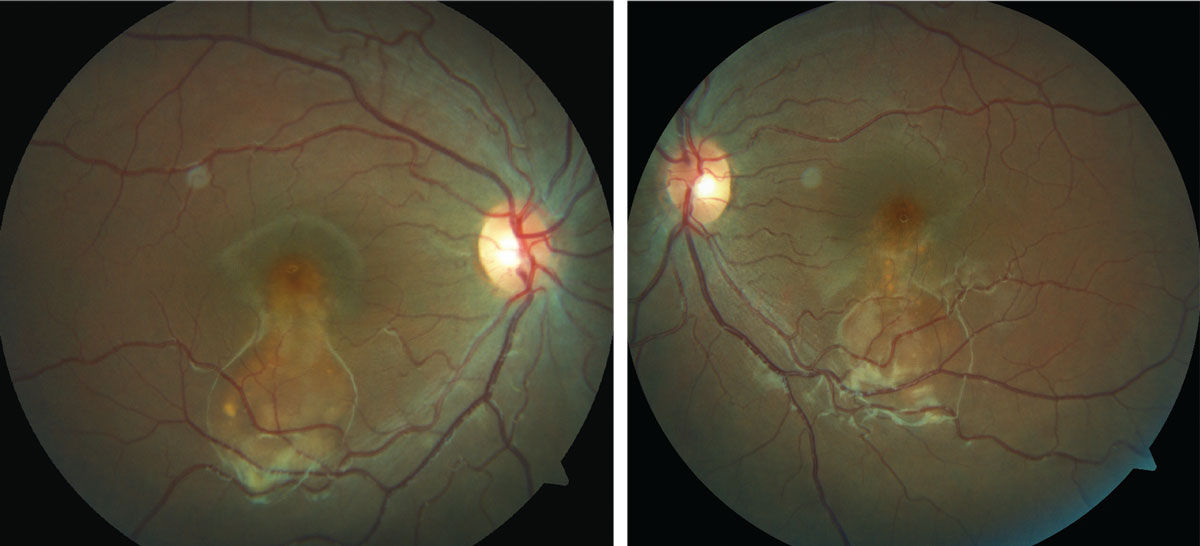 |
| This 16-year-old presented for a routine exam, but an evaluation of her fundus revealed something amiss. Can you identify it? Click image to enlarge. |
Discussion
Additional studies included refraction to determine if there was a hyperopic shift (central serous chorioretinopathy), optical coherence tomography (OCT) to examine the status of the retinal tissue and its relationship to the Bruch’s membrane and retinal pigment epithelium, optical coherence tomography angiography (OCT-A) to examine the subretinal/choroidal tissues for choroidal neovascular membrane formation and color photodocumentation. An electro-oculogram (EOG) was completed to assist with the diagnosis. A sodium fluorescein angiogram may also be indicated.
The diagnosis in this issue is atypical Best’s disease (BD). BD (or Vitelliform macular dystrophy) is an inheritable macular dystrophy described by Freidrich Best in 1905.1-6 It is an autosomal dominant macular dystrophy with a characteristic “egg-yolk” sub-retinal macular deposit known as a “vitelliform lesion”.1-6 The condition is primarily caused by mutations in the bestrophin gene which cause lipofuscin to accumulate subretinally.1-6 The classic clinical presentation is singular bilateral “vitelliform” lesions presenting in the first or second decade of lifeusually associated with good visual acuity.1-4 An adult variant known as adult vitelliform dystrophy (AVD) presents in the fourth, fifth or sixth decade and is associated with a better visual prognosis.2,3 Other findings in BD include multiple “vitelliform” lesions and serous retinal detachment.1 In the later stages of the disease the “vitelliform” material is reabsorbed leaving a scar which can confound the diagnosis.1
The natural course of the disease is divided into four stages: 1) previtelliform (normal funduscopically with hyper-reflective thickening of RPE/photoreceptor interface on OCT), 2) vitelliform (classic egg-yolk lesion), 3) vitelliruptive (mottling of vitelliform lesion with or without a pseudo-hypopyon funduscopic appearance) and finally 4) the atrophic stage.3,4,7 The evolution of the “vitelliform” lesion reflects the accumulation of lipofuscin and its eventual breakdown and reabsorption with subsequent death of the underlying RPE cells.1
The diagnosis is confirmed with electro-oculogram (EOG). The pathognomonic finding is a reduced Arden ratio (<1.5) in the setting of a normal electroretinogram (ERG).1-4 AVD differs from BD as the EOG is typically near normal although multi-focal ERG may be slightly reduced.3
Ocular coherence tomography (OCT) typically shows a dome-shaped hyper-reflective and homogenous lesion below the photoreceptor layer corresponding with the typical egg-yolk-like “vitelliform” lesion.3 Subretinal fluid and photoreceptor thinning may also be seen in the later stages of the disease. Choroidal neovascularization (CNV) may form as well.2-4 Fundus autofluorescence (FAF) typically reveals hyper-fluorescent patches corresponding with the “vitelliform” lesions.1,2,4
Vision loss is usually slow and gradual, secondary to atrophy and scarring of the macular region.3 Vision is usually preserved until the later stages of the disease but eventually succumbs to levels of 20/200.3 A sudden decrease in vision may signify the development of CNV. These typically respond well to treatment with intravitreal anti-vascular endothelial growth factor (VEGF) therapy.3,2,8,9
There are a few published case reports of BD presenting as bilateral serous retinal detachments (as occurred in this case).1,5 All published cases were originally misdiagnosed as central serous chorioretinopathy (CSCR) with correct diagnosis achieved only after photodynamic therapy (PDT) treatment failure.1,5 The differential diagnosis in these cases is broad but should include CSCR, neovascular age-related macular degeneration (wet-AMD), and polypoidal choroidal vasculopathy (PCV).1 The EOG testing is critical for correct diagnosis in these circumstances as a decreased Arden ratio is uniquely suggestive of BD.1-3
There is no treatment for BD beyond management of CNV with intravitreal injection of anti- VEGF.8,9 Home-monitoring with an Amsler grid and genetic counseling are also appropriate since there is a genetic vector.1 Low vision rehabilitation is often beneficial in advanced cases, particularly for activities involving central vision such as reading and recognizing faces.3
The patient was referred to the retinal specialist who agreed with our diagnosis and assisted in completing the EOG confirming the diagnosis. The patient was educated regarding the disease’s genetic nature and was given an Amsler grid to home monitor with the explanation to return to the office should any changes in vision begin. No additional intervention was required.
Dr. Gurwood thanks Dr. Karbach for contributing this case.
|
