 |
A 57-year-old male presented complaining of mildly decreased vision in both eyes. He noted the change a few months prior. His medical, family and social history is noncontributory. Upon examination, his best-corrected visual acuity was 20/40 OD and 20/30 OS. Extraocular motilities showed full range of motion in both eyes. His confrontation fields were full-to-careful finger counting in the right and left eye. His pupils were unremarkable with no afferent pupillary defect. Additionally, his intraocular pressures (IOP) were 17mm Hg OD and 16mm Hg OS.
Anterior segment examination was within normal limits. The fundus showed significant retinal pigment epithelium (RPE) atrophic changes in the macula of both eyes. In the right eye, we observed some elevation of the RPE. A macular optical coherence tomography (OCT) image was obtained and is available for review (Figures 1a and 1b).
Further review of the patient’s medical record showed fundus images that were taken three years prior (Figures 2a and 2b).
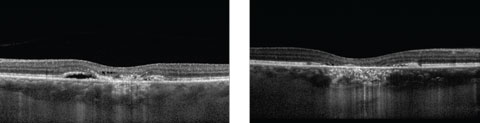 |
| Figs. 1a and 1b. These two OCT images show two stages of the patient’s condition. Can you make the diagnosis? Click image to enlarge. |
Take the Quiz
1. What is the likely diagnosis?
a. Central serous retinopathy.
b. Age-related macular degeneration.
c. Adult-onset vitelliform dystrophy.
d. Polypoidal choroidal vasculopathy.
2.What does OCT imaging show?
a. Retinal thickening with cystoid changes of the inner retina.
b. Subretinal exudates and intraretinal edema.
c. Macular edema.
d. Retinal atrophy with subretinal fluid.
3. What additional testing is warranted?
a. Fluorescein angiography.
b. Genetic testing.
c. B-scan ultrasonography.
d. No additional testing.
4. Which of the following represents the best treatment plan for this patient?
a. Observation.
b. Laser treatment.
c. Anti-VEGF.
d. b and c.
For answers, see below.
Diagnosis
Based on the fundus photos that were reviewed from three years prior, we determined that our patient has adult-onset vitelliform dystrophy. The fundus photos show a fairly classic “vitelliruptive” stage of the disease that, over the ensuing three years, has progressed to the “atrophic” stage.
Adult-onset vitelliform dystrophy is an autosomal dominant disorder with variable expression and incomplete penetrance that results in slow, progressive bilateral vision loss. The classic presentation resembles a sunny-side egg-yolk appearance, which can be appreciated in the images; however, it is apparent from the photos that the disease has progressed to the next stage—the “pseudohypopyon” stage.
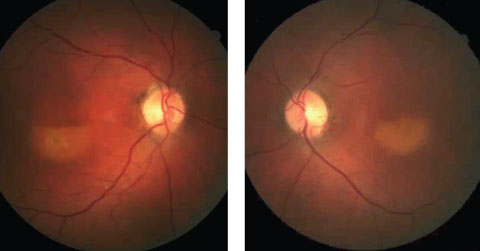 |
| Figs 2a and 2b. These fundus photos show our patient from three years prior to the diagnosis. Can this presentation, combined with the current-day OCT, lead you to a diagnosis? Click image to enlarge. |
Discussion
Adult-onset vitelliform dystrophy, initially referred to as “peculiar foveomacular dystrophy,” is divided into five distinct stages. They are:
1. Vitelliform
2. Pseudohypopyon
3. Vitelliruptive
4. Atrophic
5. Cicatricial
The “egg-yolk” lesion that presents as a yellow subretinal deposit and does not affect vision characterizes the vitelliform stage. In the pseudohypopyon stage, the yellow material liquefies, settling inferiorly and giving the appearance of a hypopyon. Acuity becomes affected once the disease progresses to the vitelliruptive stage, in which the central aspect of the lesion becomes atrophic and the “egg-yolk” takes on the “scrambled” appearance. When we most recently saw the patient, the disease had progressed to the atrophic stage in which the changes can be nonspecific. The OCT image shows disruption at the RPE level with loss at the inner segment/outer segment junction as well as in the right eye and area of subretinal fluid.
Researchers believe the disease develops as a result of genetic mutations in the photoreceptor protein-encoding genes, which leads to breakdown of the RPE-photoreceptor complex causing toxic accumulation of cellular debris at the RPE level, resulting in the classic presentation of bilateral yellow subfoveal deposits.1,2 The deposits intensify over time, ultimately resulting in RPE atrophy and vision loss.2
The age of onset remains variable with many patients remaining asymptomatic until the fifth or sixth decade of life.
Monitoring
Diagnostic testing often includes OCT, fluorescein angiography (FA) and, in some cases, electrophysiological studies. Despite variability in visual outcomes and acuity, full-field and multifocal electroretinograms in patients with adult-onset vitelliform dystrophy reveal that the disease significantly disrupts macular function.3 Furthermore, the electro-oculograms may be slightly reduced or normal.4 Early in the disease, the FA will expectedly reveal a pattern of patchy hypofluorescence at the macula with surrounding hyperfluorescence.5,6 OCT can be used to identify the anatomical location of the lesion and will reveal a linear RPE layer separated from the photoreceptor layer by cellular debris, likely to be lipofuscin.7 Note that the fluorescein pattern and OCT findings will change as the lesions progress.
As the disease evolves, vision loss becomes more severe. In the later disease stage, as visual impairment and loss of central acuity become a greater concern, patients should be issued an Amsler grid to monitor for visual changes at home. Referrals to vision rehabilitation should be made to enhance quality of life. Although rare, complications such as full thickness macular holes, choroidal neovascular membranes and retinal detachment can occur; therefore, annual dilated exams and close observation are warranted.8
Dr. Avdic is a resident at Bascom Palmer Eye Institute in Miami.
| 1.Felbor U, Schilling H, Weber B. Adult foveomacular vitelliform dystrophy is frequenty associated with mutations in the peripherin/RDS gene. Hum Mutat 1997;10:301-9. 2.Chowers I, Tiosano L, Audo I, Grunin M. Adult onset foveomacular vitelliform dystrophy: A fresh perspective. Prog Retin Eye Res. 2015:47:64-85. 3. Yamamoto S, Saito W, Ogata K, Hayashi M. Electrophysiologic studies on patinets with adult onset vitelliform macular degeneration. Invest. Ophthalmol. Vis. Sci. 2002;43(13):1162. 4. Birndorf L, Dawson W. A normal electrooculogram in a patient with a typical vitelliform macular lesion. Invest Ophthalmol. 1973:12(11):830-3. 5. Parodi M, Iacono P, Campa C, et al. Fundus Autofluorescence Patterns in Best vitelliform macular dystrophy. Am J Ophthalmol 2014;158(5):1086-92. 6. Pierro L, Tremolada G, Introlini U, et al. OCT findings in adult onset foveomacular vitelliform dystrophy. Am J Ophthalmol 2002;134:675-80. 7. Benhamou N, Messas-Kaplan A, Cohen Y, et al. Adult onset foveomacular vitelliform dystrophy with OCT. Am J Ophthalmol 2004;138(2):294-6. 8. Tiosano L, Grunin M, Hagbi-Levi S, et al. Characterizing the phenotype and progression of sporadic adult onset foveomacular vitelliform dystrophy. Br J Ophthalmol 2016;100:1476-81. |
Retina Quiz Answers:
1) b; 2) d; 3) a; 4) a.
