In April 2016, optometry lost a giant when the author of the seminal work Primary Care of the Posterior Segment, Larry Alexander, OD, died. In addition to being an optometric physician, author and educator at the University of Alabama Birmingham School of Optometry, Dr. Alexander was a past president of the Optometric Retina Society (ORS). That group chose to honor his legacy by accepting case reports from optometric residents across the country relating to vitreoretinal disease.
This case, selected by the ORS Awards Committee, was co-winner of the sixth annual Larry Alexander Resident Case Report Contest. The contest is sponsored by Topcon, Visionix (Optovue) and Heidelberg.
Multifocal vitelliform dystrophy (MVD) is a clinically and genetically heterogeneous retinal disease that presents as bilateral multiple yellow lesions within the posterior pole, which is caused by subretinal accumulation of vitelliform material. The age of onset is highly variable, ranging from five to 59 years old, and common symptoms include blurry vision and central metamorphopsia.1 Previously reported cases of multifocal vitelliform lesions have been described as raised yellow lesions located near the macula, along the retinal vascular arcades or adjacent to the optic nerve.1 The condition’s etiology may be familial or sporadic,2 and can be associated with a mutation in the bestrophin 1 (BEST1) gene. MVD with mutations in BEST1 gene can be referred to as Multifocal Best disease (MBD), which is a rare phenotype of Best vitelliform macular dystrophy.
Diagnosis of MVD is mainly based on clinical presentation and ancillary testing such as optical coherence tomography (OCT), fundus autofluorescence (FAF), fluorescein angiography (FA), genetic testing and electrooculography (EOG). An OCT performed over the lesion will typically show subretinal vitelliform material accompanied by adjacent subretinal fluid. FAF will usually demonstrate hyper-autofluorescence due to the accumulated lipofuscin within the lesions. FA can have variable presentations with early stages showing hypo-fluorescence and late stages showing hyper-fluorescence. Some studies showed that most cases of MVD have partial staining on fluorescein angiography.2-4 An EOG can demonstrate a reduced response but most patients with multifocal vitelliform lesions exhibit a normal EOG.2,5,6 MVD can be monitored without treatment; however, there are secondary complications like choroidal neovascular membrane that can be vision-threatening. Clinicians must monitor for secondary complications of MVD and appropriately refer out cases in need of urgent treatment.
This case report highlights some of the unique clinical features of MVD, discusses how to differentiate this rare condition from other retinal conditions that may have similar presentations and recommends proper clinical management.
Case report
|
Check out the other winning submission of the ORS resident case report contest on bilateral Bartonella henselae neuroretinitis. |
A 67-year-old male presented for a comprehensive eye exam with a chief complaint of blurry vision at near in both eyes without glasses. His ocular history was significant for pigmentary epithelial detachments in both eyes and a dendriform pattern corneal scar of the right eye likely due to history of herpes simplex virus keratitis. The patient’s medical history was notable for hypertension, hyperlipidemia, prediabetes and depression. His current medications were lisinopril, hydrochlorothiazide, metoprolol, nifedipine, Lipitor, citalopram, trazodone and Vistaril. The patient reported no known drug allergies and an unremarkable family ocular history.
The patient’s best-corrected visual acuity was 20/40-2 OD and 20/40 OS with a refractive error of +1.50 -0.50 x 80 ADD +2.50 OD and +1.75 -0.75 x 155 ADD +2.50 OS. Pinhole testing showed no improvement in vision of either eye. Pupils were round equal, reactive to light with no afferent pupillary defect in either eye. Extraocular motilities were full OU, and confrontation visual fields were full to finger counting OU.
 |
|
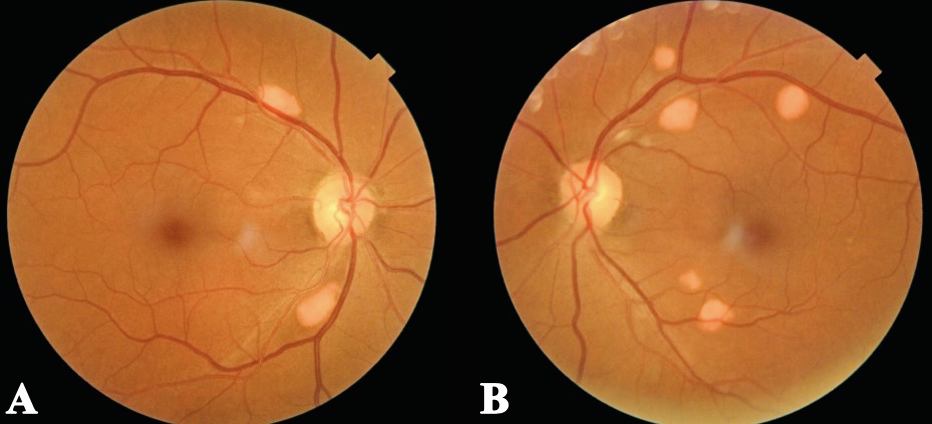
Figure 1. Fundus photos of both eyes show scattered elevated, orange-yellow lesions with distinct borders, located along the superotemporal and inferotemporal retinal vascular arcades within the posterior pole. Click image to enlarge. |
Anterior segment examination was unremarkable besides the dendritic corneal stromal scar OD. Intraocular pressure was normotensive OU. Posterior segment examination revealed scattered orange-yellow discrete lesions along the superior and inferior temporal arcades OU in the posterior pole. These lesions were elevated with distinct borders and fundus photos were taken for documentation (Figure 1).
According to the patient’s previous records from four years ago, these orange-yellow lesions were longstanding but appeared to be changing over time (these lesions were first noted OS and then progressed to OU). No previous fundus photo documentation or detailed description was available to compare the lesion numbers, size, location and appearances. The patient was asymptomatic, but given the suspicion for progression, further testing such as a macula OCT through the posterior pole, FAF, FA and genetic testing was warranted.
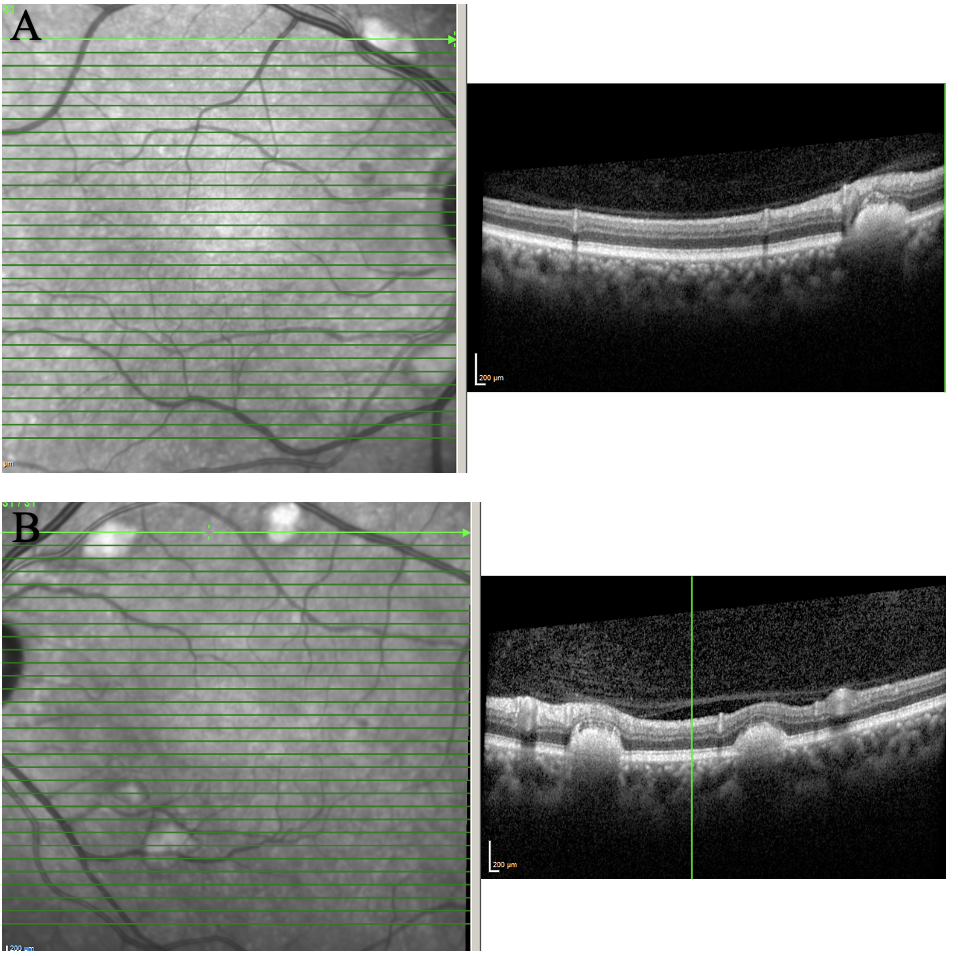 |
| Figure 2. OCT of the posterior pole of OD (A) and OS (B) showing scattered subretinal deposits along the arcades. The lesions were elevated and caused outer retinal layer disruption and elevation. Click image to enlarge. |
OCT of posterior pole (Figure 2) showed scattered subretinal deposits along the arcades. These lesions were raised and caused outer retina layer disruption and elevation. There was no choroidal neovascular membrane or outer retina atrophy. Maculas of both eyes were uninvolved and remained flat and intact.
 |
|
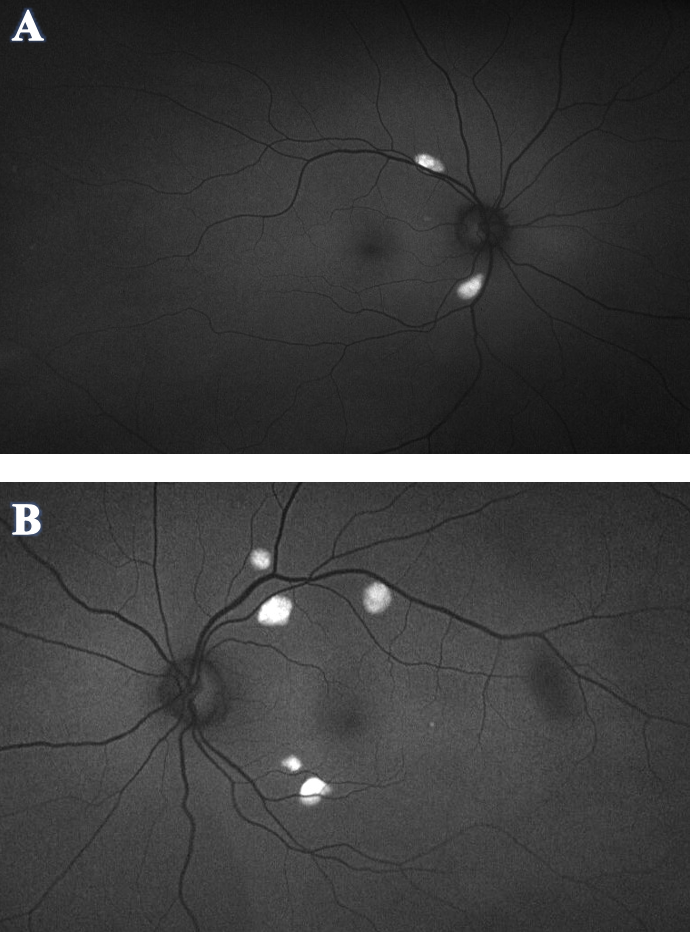
Figure 3. Optos FAF of OD (A) and OS (B) demonstrated hyper-autofluorescence corresponding to the orange-yellow lesions seen in the fundus photos. No hypo-autofluorescence was appreciated in either eye. Click image to enlarge. |
Optos FAF (Figure 3) demonstrated hyper-autofluorescence corresponding to the orange-yellow lesions in fundus photo. No hypo-autofluorescence was appreciated in either eye.
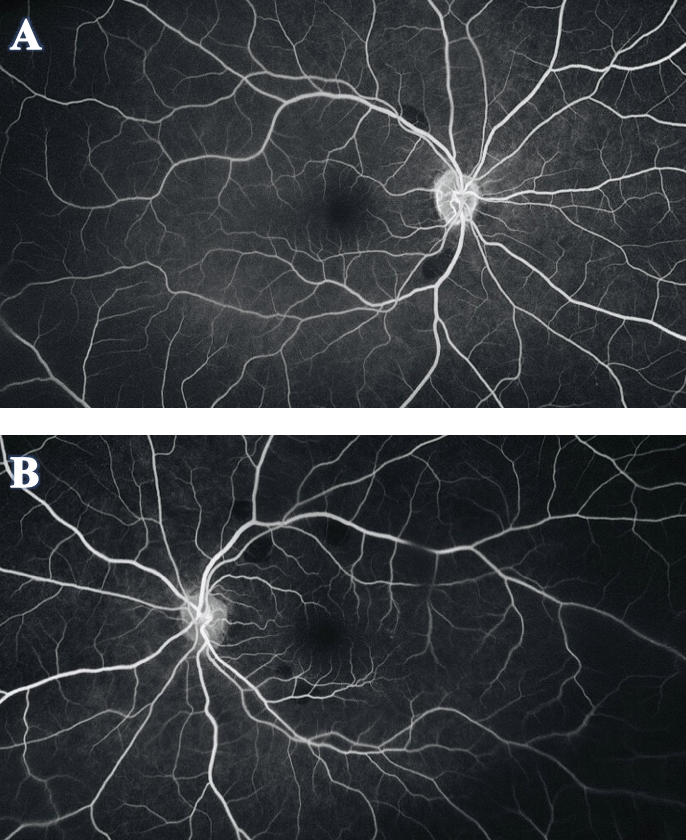
To further investigate the etiology of the lesions, FA was recommended (Figure 4). The scan showed that the orange-yellow lesions corresponded to areas of hypo-fluorescence or a blockage of fluorescein. There was no early- or late-phase leakage or staining, and normal retinal perfusion was observed in both eyes.
The patient’s B-scan (Figure 5), as well as the A-scan, demonstrated that the orange-yellow retinal lesions were hyper-echogenic and hyper-reflective flat lesions with distinct borders located within the posterior pole. Most of the lesions were observed along the retinal vascular arcades or near the optic nerve. An OCT scan through these retinal lesions showed subretinal deposit of vitelliform material that hyper-autofluorescent on FAF and hypo-fluorescent on FA.
Genetic testing was recommended to evaluate for any inherited retinal dystrophies. The patient denied any family history of ocular conditions or issues with night vision. The type of genetic testing used was Invitae Inherited Retinal Disorders Panel, which produced a result of “uncertain,” with variants of uncertain significance identified. The testing confirmed that there was no gene variation or mutation for the BEST1 gene or evidence of other inherited retinal disorders, indicating that MBD could be ruled out as a possible diagnosis.
Differential diagnosis
Sclerochoroidal calcification is a condition caused by deposition of calcium in the sclera that leads to a flat or elevated choroidal or scleral mass commonly mistaken for an ocular tumor.7-9 It can have a similar appearance to MVD: both present as raised yellow lesions in the retina. However, this differential was ruled out in this patient’s case since the OCT demonstrated that the lesions were subretinal with retinal pigment epithelium (RPE)-Bruch's membrane complex intact, which indicates that they were not coming from the choroid or sclera. There was also an absence of choroidal or scleral thickening that is typically present in cases with sclerochoroidal calcification.
 |
|
Figure 4. FA of OD (A) and OS (B) showed the orange-yellow fundus lesions corresponded to areas of hypo-fluorescence or a blockage of fluorescein. There was no leakage or staining in early or late phase, and normal retinal perfusion was observed in both eyes. Click image to enlarge. |
The B-scan of the orange-yellow lesions in this case of MVD was hyper-echogenic, which also resembles choroidal osteoma—a rare, benign tumor that often presents as unifocal, unilateral yellow-white lesion that can increase in size and decalcify over time.10 Choroidal osteoma was ruled out based on the difference in OCT presentation: there was no choroid thickening, and the lesions were subretinal; in addition, the presentation was bilateral, small, multifocal lesions, whereas choroidal osteoma tends to consist of a unilateral, unifocal, large lesion.
Another differential to be considered in these patients is autosomal recessive bestrophinopathy, which can have a very similar presentation as MVD, with multifocal yellowish subretinal lesions associated with hyper-autofluorescence on FAF.11 Genetic testing of these patients will show a mutation in the BEST1 gene as null phenotype where both alleles must be mutated.12 There are also additional ocular abnormalities associated with this condition: hyperopia, amblyopia, angle closure glaucoma and short axial length.13 This disease was ruled out by genetic testing and the absence of additional ocular abnormalities that are typically found in autosomal recessive bestrophinopathy.
Acute exudative polymorphous vitelliform maculopathy, like the name suggests, has an acute onset characterized by local serous retinal detachment then followed by development of yellow-white lesions.14 This was ruled out based on the difference in time of onset (acute vs. chronic) and absence of subretinal fluid. Paraneoplastic acute exudative polymorphous vitelliform maculopathy was also ruled out based on patient had no history of malignancy.
Age-related macular degeneration can present as sub-RPE lesions on OCT. This was ruled out based on absence of drusen in the patient’s fundus especially in the macular region or in the retina surrounding the subretinal lesions where drusen are typically found.
Multifocal serous detachment of the RPE is another condition that can have similar fundus appearance compared to MVD. This condition was ruled out based on the absence of fluid on the OCT.
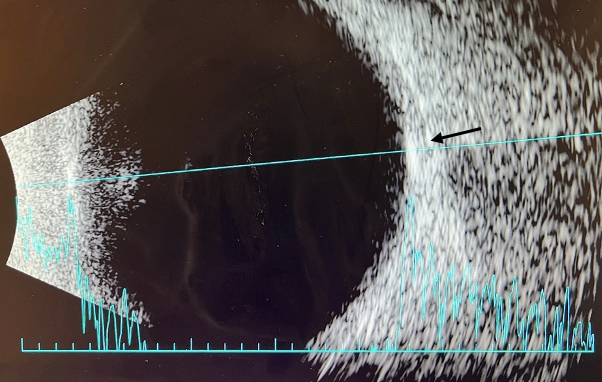 |
| Figure 5. B-scan of right eye. A B-scan was performed on the right and left eye with similar findings in both. Retinal lesions were noted to be hyper-echogenic and hyper-reflective flat lesion on A and B scan (arrow). Click image to enlarge. |
Scattered yellow lesions in the retina can also be secondary to inflammatory etiologies. Chorioretinitis like bird shot chorioretinitis and multiple evanescent white dot syndrome were ruled out based on the absence of ocular inflammation (no vitritis or vasculitis).
Lymphoma was ruled out because the patient had no history of malignancy, an unremarkable complete blood count and no lesions appeared to be chronic. The patient also did not have a rapid and acute onset, which is often the case with lesions that are due to lymphoma.
Discussion
The 67-year-old male featured in this case report had multiple bilateral, distinct, orange-yellow lesions along the retinal vascular arcades within the posterior pole. The patient was asymptomatic, and based on previous records, the lesions appeared to be chronic and progressive over time. FAF demonstrated increased autofluorescence correlating to the orange-yellow lesions, and OCT scans over the lesions showed subretinal accumulation of vitelliform material that resembled MVD. FA showed hypo-fluorescence of the lesions during early and late phases with no staining or leakage, which can be attributed to the blocking effect caused by the vitelliform lesions. The patient denied any family history of ocular disease, and family members were not examined in this case. The diagnosis of MVD was based on clinical appearance, negative genetic testing, OCT findings of subretinal vitelliform materials, hyper-autofluorescence on FAF and hypo-fluorescence on FA. EOG in this case would also aid in the diagnosis, however, was deemed not necessary because previous literature has shown that EOG tends to be normal or mildly subnormal in patients with MVD and MBD,2 especially in those without genetic mutations.1
The difference between MVD and MBD is that patients with MVD lack the genetic mutation of BEST1 gene. Boon et al. studied patients who have this rare phenotype of multifocal vitelliform lesions and further investigated the difference in clinical features in patient groups with and without genetic mutations in BEST1 gene. Fundus appearance, OCT, FAF and FA findings are similar between the two groups, but they discovered that lesions in patients without the genetic mutation tend to be smaller in size and higher in numbers. EOG findings were normal in four out of six patients without the genetic mutation.
In comparison to the patient group carrying the genetic mutations, patients without BEST1 gene variations tend to have an older age of onset, less affected visual acuity and normal EOG findings.1 Abnormal EOG findings in patients carrying the mutation was expected since mutations in BEST1 gene correlated to more extensive RPE dysfunction rather than an abnormal EOG.15 Normal EOG findings in patients without the genetic mutation suggested there is no broad RPE electrical activity dysfunction.1 Apart from the difference in clinical features, the absence of mutation in BEST1 gene does not necessarily mean patients were lacking the disease-causing gene variation since genetic tests carry a chance of false negativity. There was also a possibility that heterozygous deletion of exons was missed by the polymerase chain reaction. Another explanation would be the involvement of an unknown gene in the pathophysiology of those multifocal vitelliform lesions that has not yet been discovered and is not in the current database.1 Hence, it would be more prudent to describe this case presentation of multifocal vitelliform lesions in the absence of BEST1 mutation as MVD.
The underlying pathophysiology for multifocal vitelliform lesions is unknown. In MBD, it has been suggested that the mutation in BEST1 gene has caused ion channel dysfunction in the protein leading to ionic imbalance in the RPE milieu and eventually RPE dysfunction.16 The accumulation of subretinal lipofuscin has also been suggested as part of the pathophysiology, although some studies suggested it’s more of a sign of premature RPE dysfunction.16,17 Multifocal vitelliform lesions can also present in other differential diagnoses like acute exudative polymorphous vitelliform maculopathy and paraneoplastic acute exudative polymorphous vitelliform maculopathy. These conditions were ruled out in this patient based on differences in clinical presentation, clinical history and using multimodal retinal imaging. A case report of multifocal vitelliform lesions in Alport Syndrome suggested a potential link between structural changes in the type IV collagen and formation of subretinal vitelliform lesions due to the underlying disruption of Bruch’s membrane.18 However, multifocal vitelliform lesions in Alport syndrome is rare, whether this is a true association or isolated finding still awaits further investigation.
There is no current effective medical or surgical treatment to alleviate or cure MVD; therefore, clinical management of the disease consists largely of monitoring without intervention. Patients need to be monitored every four to six months with dilated fundus exams and OCT scans, and doctors also need to be attentive for incidence of secondary complications such as choroidal neovascular membrane, RPE atrophy, sub-RPE fibrosis and geographic atrophy.19 In the case of a choroidal neovascular membrane, patients need to be referred to a retinal specialist promptly for potential treatment such as laser, photodynamic therapy or intravitreal injection of anti-VEGF.20-22 Clinicians should also provide detailed patient education on signs and symptoms of acute visual changes and to return to clinic urgently for evaluation if these occur. An Amsler grid can be dispensed for patients to self-monitor at home in between scheduled follow-up appointments. In cases where central vision is affected or impaired, referral to a low vision clinic should be warranted. Clinicians should also consider recommending ocular examination and genetic testing to the family members of patients with MVD.
Conclusion
MVD usually presents as orange-yellow colored subretinal deposits located within the posterior pole. It can be associated with BEST1 gene variation (referred to as MBD) but not present in all cases.1 Since MVD is a rare phenotype, other etiologies like inflammatory, infectious and neoplastic conditions need to be ruled out by ocular imaging and lab work. Visual impairment in MVD can vary from case to case depending on the level of central vision involvement. There is no current treatment for MVD, and secondary complications including choroidal neovascular membrane warrant a prompt referral to a retinal specialist for management. Due to its low prevalence, there are currently limited studies on MVD. Future similar encounters may provide additional insight on etiology, pathophysiology, treatment and management of this vision-threatening retinal disease.
Dr. Keying would like to thank her attending ophthalmologist, Zimei Zhou MD, PhD, for co-managing this case, as well as her attending optometrists, Alanna Khattar-Sullivan, OD, and Tybee Eleff OD, for helping with editing this case report.
1. Boon CJ, Klevering BJ, den Hollander AI, et al. Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol. 2007;125(8):1100-1106. 2. Lisch W, Weidle EG, Richard G, et al. Multiple vitelliform retinal cysts: case report and review of the literature. Klin Monatsbl Augenheilkd. 1989;194(2):120-128. 3. Denden A, Littann KE. Fluorescent angiography in multiple vitelliform retinal cysts of the posterior ocular pole. Ophthalmologica. 1973;166(2):94-104. 4. Miller SA. Multifocal Best's vitelliform dystrophy. Arch Ophthalmol. 1977;95(6):984-990. 5. Pollack K, Kreuz FR, Pillunat LE. Best's disease with normal EOG. Case report of familial macular dystrophy. Ophthalmology. 2005;102(9):891-94. 6. Hittner HM, Ferrell RE, Borda RP, et al. Atypical vitelliform macular dystrophy in a 5-generation family. Br J Ophthalmol. 1984;68(3):199-207. 7. Shields JA, Shields CL. CME review: Sclerochoroidal calcification: The 2001 Harold Gifford Lecture. Retina 2002;22:251-61. 8. Shields JA, Shields CL. Intraocular Tumors: An Atlas and Textbook. 2nd ed. Philadelphia, PA. Lippincott Williams and Wilkins. 2008:194. 9. Fung AT, Arias JD, Shields CL, et al. Sclerochoroidal calcification is primarily a scleral condition based on enhanced depth imaging optical coherence tomography. JAMA Ophthalmol. 2013;131:960-63. 10. Alameddine RM, Mansour AM, Kahtani E. Review of choroidal osteomas. Middle East Afr J Ophthalmol. 2014;21(3):244-50. 11. Boon CJ, van den Born LI, Visser L, et al. Autosomal recessive bestrophinopathy: Differential diagnosis and treatment options. Ophthalmology. 2013;120(4):809-20. 12. Johnson AA, Guziewicz KE, Lee CJ, et al. Bestrophin-1 and retinal disease. Prog Retin Eye Res. 2017;58:45-69. 13. Burgess R, Millar ID, Leroy BP, et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82(1):19-31. 14. Barbazetto I, Dansingani KK, Dolz-Marco R, et al. Idiopathic acute exudative polymorphous vitelliform maculopathy: clinical spectrum and multimodal imaging characteristics. ophthalmology. 2018;125(1):75-88. 15. Marmorstein AD, Kinnick TR. Focus on molecules: Bestrophin (best-1). Exp Eye Res. 2007;85(4):423-24. 16. Guziewicz KE, Sinha D, Gómez NM, et al. Bestrophinopathy: An RPE-photoreceptor interface disease. Prog Retin Eye Res. 2017;58:70-88. 17. Singh R, Shen W, Kuai D, et al. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet. 2013;22(3):593-607. 18. Eriksen KO, Jørstad ØK. Multiple vitelliform lesions as a retinal manifestation of Alport syndrome. Case Rep Ophthalmol. 2020 Feb 12;11(1):79-84. 19. Schachat AP, Wilisoson CP, Hilton DR, et al. Ryan’s Retina. 6th ed. Elsevier. 2018. 20. Ozdek S, Ozmen MC, Tufan HA, et al. Photodynamic therapy for best disease complicated by choroidal neovascularization in children. J Pediatr Ophthalmol Strabismus. 2012;49(4):216-21. 21. Andrade RE, Farah ME, Costa RA. Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization in best disease. Am J Ophthalmol. 2003;136(6):1179-81. 22. Khan KN, Mahroo OA, Islam F, et al. Functional and anatomical outcomes of choroidal neovascularization complicating BEST1-related retinopathy. Retina. 2017;37(7):1360-70. |
