A wide range of systemic diseases have ocular manifestations, sometimes presenting as the first or only sign of underlying illness. Crystalline keratopathy, though rare, is a corneal condition that could be signaling the presence of a number of systemic diseases, infections, disorders, adverse medication effects, cases of plant toxicity, ocular issues or other related causations (Table 1). The condition is defined as the deposition of crystals in the corneal epithelium, stroma and/or endothelium.1
The appearance of crystalline keratopathy in the cornea ranges from needle-shaped refractile polychromic crystals to dust-like, glittering granules. Imaging and observation techniques alone will not reveal the condition’s culprit; rather, identification of the underlying cause of crystalline keratopathy will require a medical workup, including laboratory testing, biopsies and/or elimination of etiologies. This case report explains how to detect some of the condition’s possible etiologies by describing the unique clinical signs, ocular manifestations and management options available for affected patients.
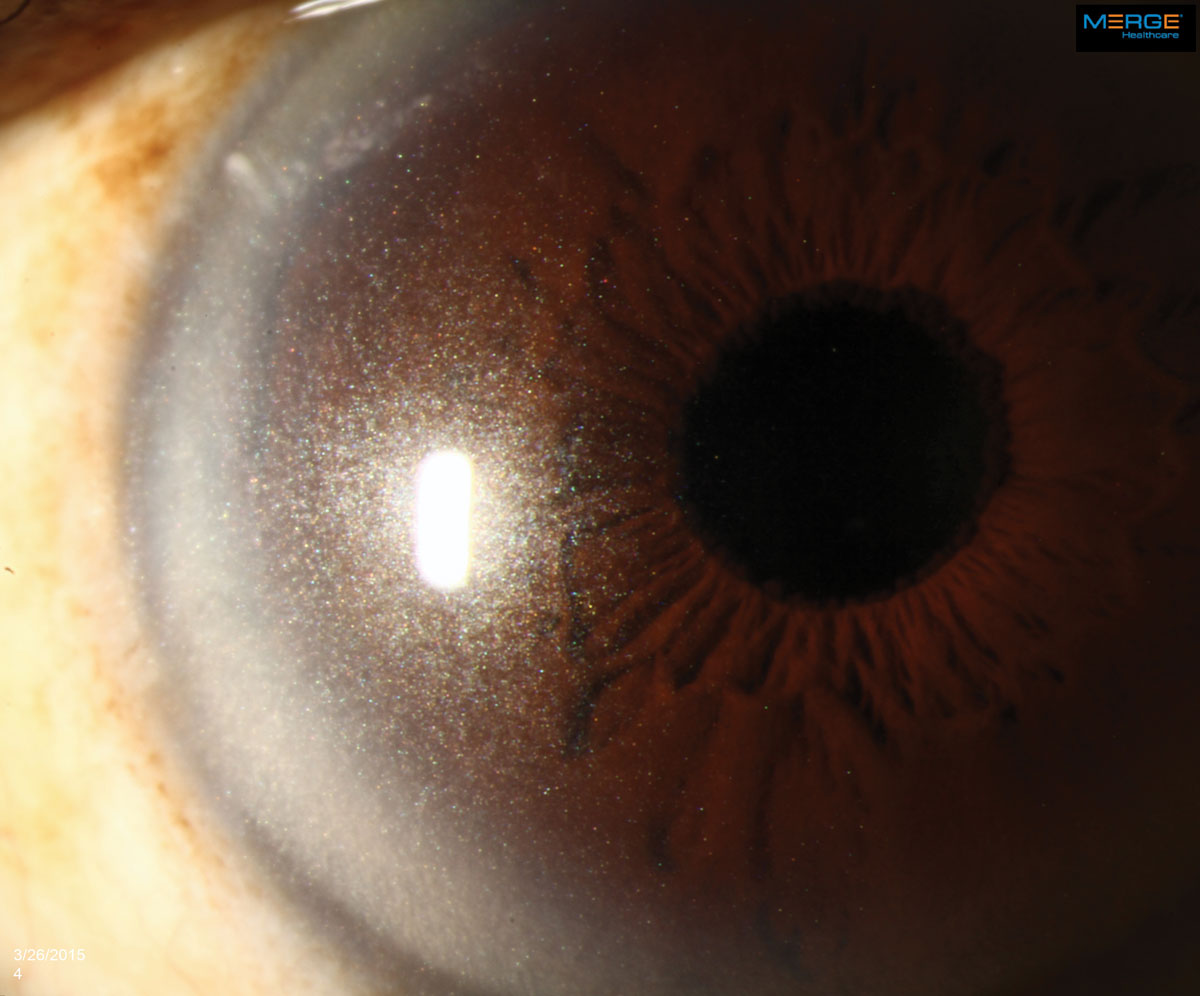 |
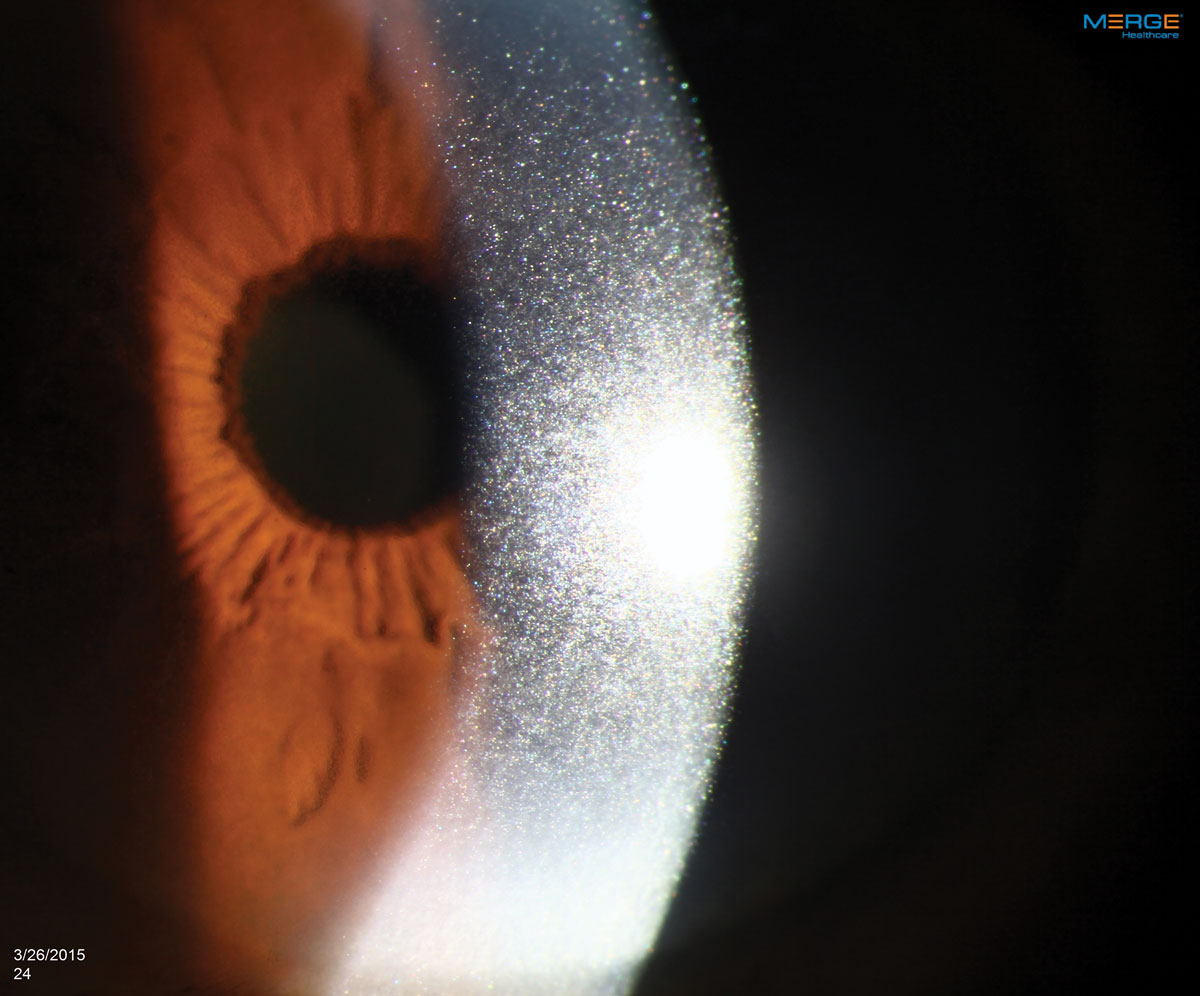
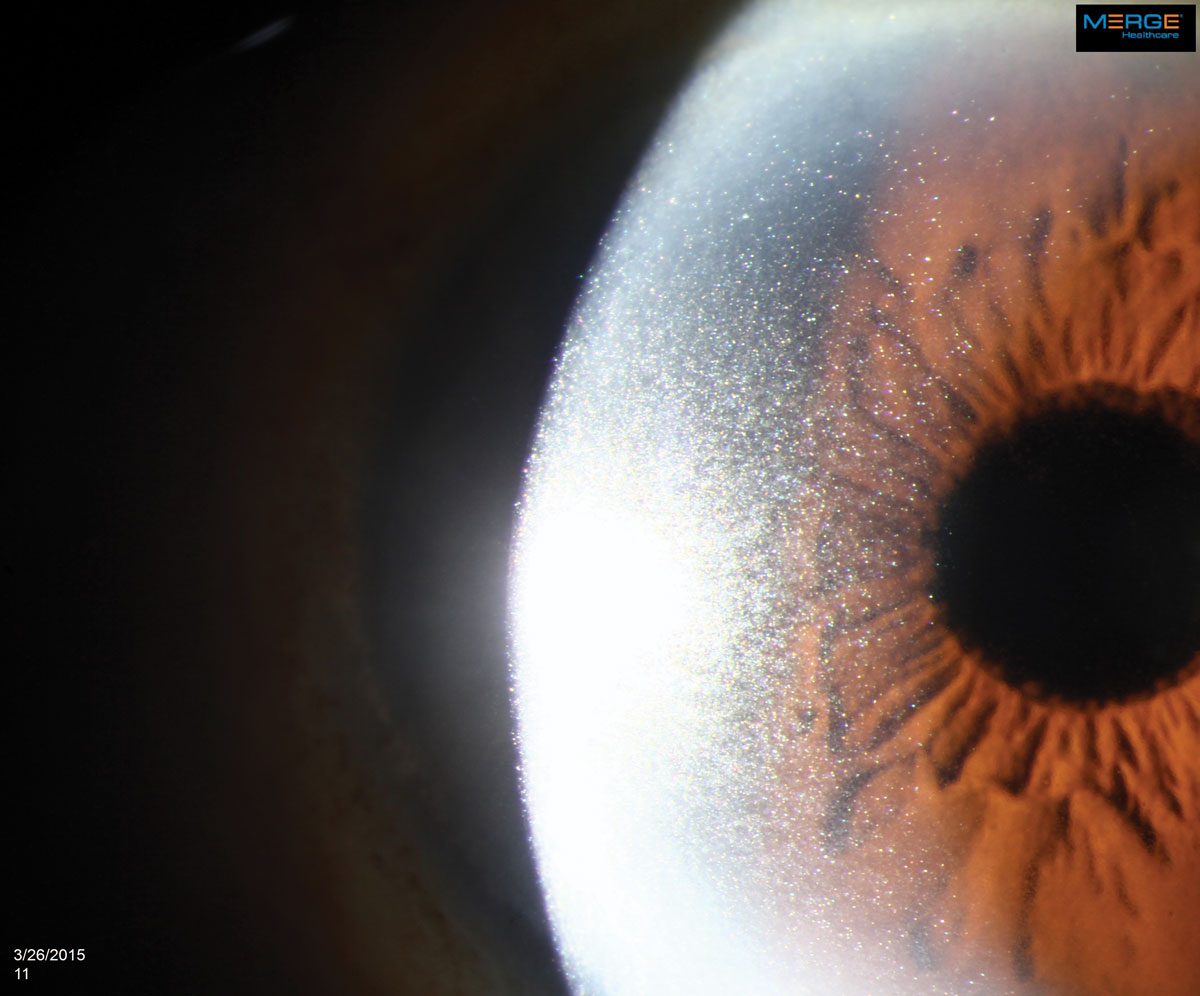
The appearance of crystalline keratopathy in the cornea can range from looking like needles to small crystals or dust-like particles. Click image to enlarge. |
Case Report
An 84-year-old African-American male presented for a comprehensive examination. His previous ocular history was remarkable for bilateral uncomplicated cataract surgery five years ago, and his best-corrected visual acuities measured 20/20 in both eyes at distance and near. His external examination was normal and there was no afferent defect. His refraction revealed stable hyperopia with presbyopia. Biomicroscopy uncovered dense, bilateral, iridescent and polychromatic deposits within the full thickness of the corneal optic section from limbus to limbus. There was a greater concentration centrally. Intraocular pressures measured 16mm Hg, OU. Dilated fundus examination revealed normal posterior pole findings with no peripheral pathologies.
A cornea specialist on staff at Scheie Eye Institute (Stephen Orlin, MD) advised contacting the primary immediately to request the patient be worked up for multiple myeloma based on his age and previous laboratory results. The other differentials (e.g., infection, toxic reaction, genetic inherited diseases) did not fit his age and he had no recent ocular surgery.
The crystalline keratopathy was photo-documented, and the patient was informed that laboratory work would be ordered through correspondence with his medical provider. Correspondence was sent explaining the finding of crystalline keratopathy and its common etiologies with recommendations for systemic laboratory work. A one-month follow-up ophthalmic appointment was scheduled to ensure follow through and complete a reevaluation ensuring condition stability.
The laboratory testing was positive for anemia, hypercalcemia, elevated creatinine and elevated serum total protein. Urinalysis results demonstrated elevated levels of total urine protein, urine creatinine, urine microalbumin and microalbumin/creatinine levels. Based on the laboratory results, the general practitioner, in collaboration with a hospital oncologist, arranged for hospital admission with scheduling of neuroimaging and a bone marrow biopsy. The bone marrow biopsy revealed IgG/kappa plasma cell myeloma. Overall test results were consistent with the diagnosis of stage III IgG kappa multiple myeloma with normal cytogenetics.
The patient was enrolled for a 28-day cycle of treatment with cyclophosphamide, bortezomib and dexamethasone (CyBorD). Acyclovir was added prophylactically for herpes zoster protection. The oncologist deferred the use of plasmapheresis to reduce the kappa light chains based on the patient’s frail condition. Chemotherapy successfully reduced kappa light chains, and his vision and ocular comfort remained unaffected. Over the course of his systemic treatment, biomicroscopy revealed a reduction in the density of corneal crystal deposition.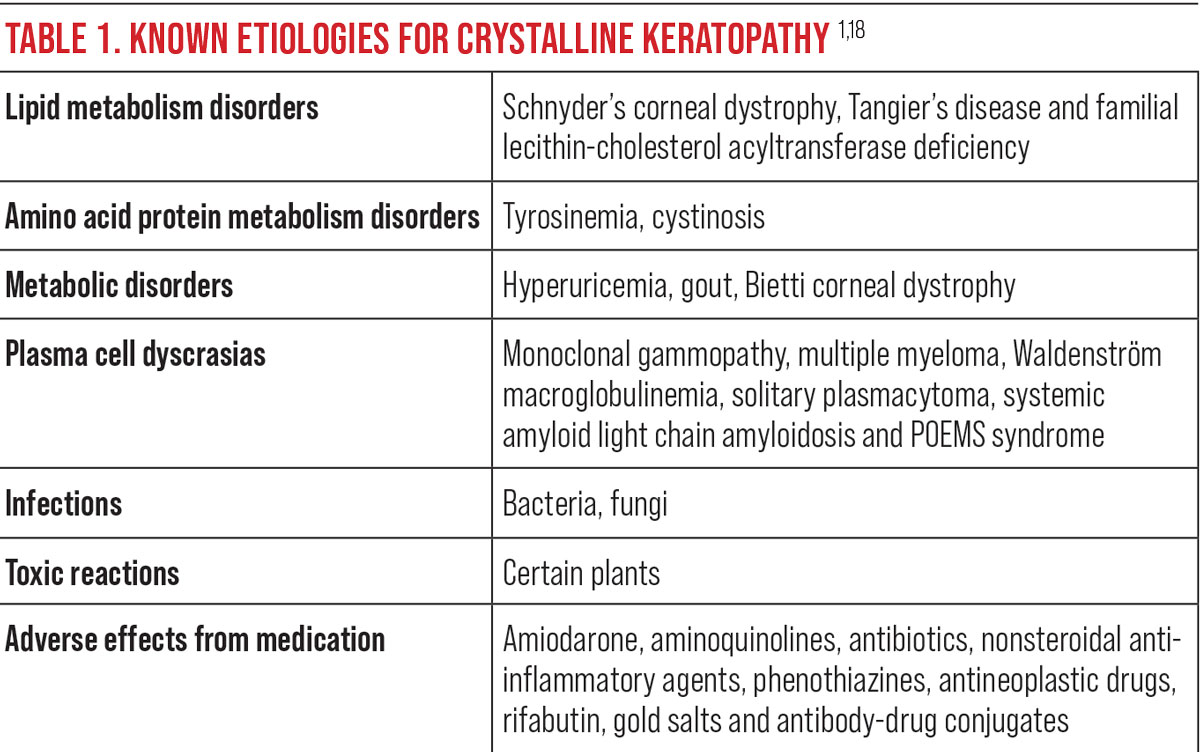
Discussion
Multiple myeloma (MM) is a malignant plasma cell dyscrasia in which clonal proliferation of B lymphocytes causes excessive levels of a single immunoglobulin referred to as a monoclonal or M-protein.18-20 MM is the most common plasma cell malignancy, accounting for approximately 10% of hematological malignancies and 1% of all cancers.19-22 The annual incidence is four per 100,000 people, with 85% diagnosed over age 65. It is more common in men than women and twice as common in the African race compared with Cacausians.18-23 In fact, MM is the most common hematologic malignancy among Blacks in the United States, with a mean age of diagnosis four years earlier than that of whites.23
Monoclonal gammopathy of undetermined significance (MGUS) is an asymptomatic premalignant variation of MM defined by elevated serum or urine protein.22 MGUS occurs in 3% to 4% of the population over age 50, with over 50% having the condition for as many as 10 years prior to diagnosis.22 MGUS can be diagnosed with routine laboratory testing. Its occurrence is two times higher in African-Americans than in Caucasians.20-23 Patients with MGUS progress to MM at a rate of 1% per year.20-23,26 Twenty percent of patients diagnosed with MGUS convert to myeloma, amyloid or lymphoma within 20 years, and 70% of those patients monitored with periodic laboratory testing develop MM.21,22,25-27
‘Smoldering MM’ is a designated term for cases in the advanced asymptomatic premalignant phase.22,28 It has a variable rate of progression to malignancy; 10% per year for the first five years, 3% per year for the next five years and 1.5% per year after that.21,22,25,28 Those identified with high-risk cytogenetic abnormalities progress more rapidly, having up to a 50% risk of malignancy within two years.22,28
Symptoms of MM include bone pain, fatigue, weakness and weight loss, with characteristic clinical findings of hypercalcemia, anemia, renal insufficiency and lytic bone lesions with increased risk of infection.19,21,22,25,27 Imaging studies show diffuse osteoporosis and/or lytic lesions of the red marrow-bearing bones including the skull, spine, ribs, proximal extremities and pelvis. This increases the risk of pathological fractures in these bones.18,19,22 Increased numbers of plasma cells can also be observed upon bone marrow biopsy.18,19,22 Experts use fluorescent in situ hybridization (FISH) probes for cytogenetic classification to identify those with high-risk abnormalities.18,19,22
Ocular manifestations of MM are rare but have been reported in every ocular structure except the lens.18,19,29-31 Crystalline keratopathy occurs in less than 1% of gammopathy cases and results from the crystallization of the M-protein within the cornea.19,30,31 The deposits are predominantly from IgG kappa light chains.19,30-32 The crystals themselves are benign but have the potential, as they amass, to cause decreased vision by altering the optics of the cornea, producing pain and photophobia through inflammatory pathways. They may also remain without causing any symptoms.27,32-34 Their widely variable presentation of color, shape, appearance, layer of cornea involved and pattern of distribution can mimic common corneal dystrophies inciting ineffective clinical managements, delaying medical testing capable of identifying the proper systemic diagnosis.2,5,16,18,30
Though the etiology of the depositions is not certain, proposed mechanisms include immunoprotein transport via the tear film, diffusion from aqueous humor, increased permeability of limbal vessels and synthesis within the keratocytes.26,34,35
Confocal microscopy studies have described the crystal shapes as granular, globular and needle-shaped in appearance.17,18,32,33 The typical shape identified on electron microscopy has been hexagonal with central cores and tubulars with a central lucent core.34,35
The immunoglobulin deposits have a strong affinity to draw copper into the cornea, further contributing to corneal irregularities and visual degradation.30,31 Central circular yellow-brown discoloration of the cornea results from pigmentation of Descemet’s membrane, and the copper deposition may extend to the lens capsule. This deposition pattern is different from the annular copper deposits found peripherally in Descemet’s membrane in those with Wilson’s Disease.31
Ciliary body cysts are associated with MM and occur in 33% to 50% of patients.19,29,30 Most are not detected on clinical examination due to location and transparency.19,29,30
Retinal findings associated with MM include microaneurysms, cotton wool spots, dilated veins, intraretinal hemorrhages, Roth spots, vein occlusions and optic disc edema (all of which should demand an order for laboratory work to screen for diabetes, hypertension, coagulopathy, hyperviscosity, dyslipidemia, cardiac, carotid and giant cell etiologies).19,29,30
Neuro-ophthalmic manifestations include diplopia from plasma cell infiltration of the ocular motor nerves, proptosis resulting from plasmacytomas in the soft tissue or orbital bones and vision loss from optic nerve involvement.19,30
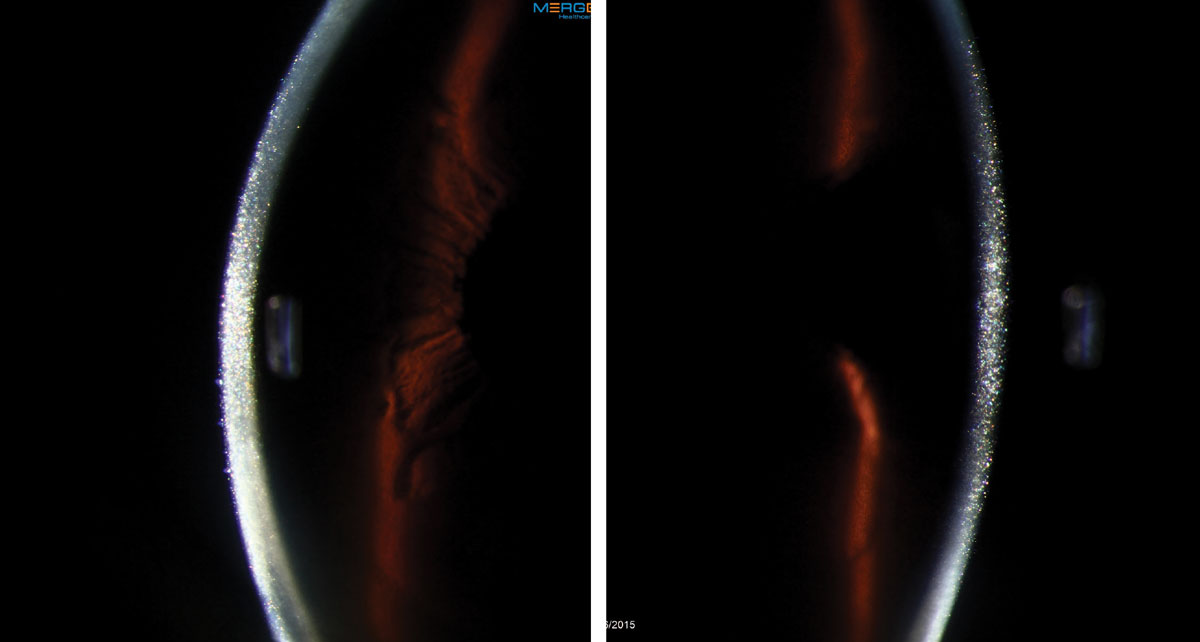 |
This 56-year-old female had a persistent epithelial defect secondary to trichiasis (caused by squamous cell carcinoma lid surgery). She wore a bandage contact lens for pain and developed an ulcer. Click images to enlarge. |
Differential Diagnosis
Since crystalline keratopathies may originate from other etiologies, these must be ruled out.1-5,36 Infectious crystalline keratopathy (ICK) is a slowly progressive discrete keratitis characterized by needle-like branching opacities. ICK does not occur as a primary disease; it results as a complication after an epithelial defect from injury or surgery and is always symptomatic. ICK has been reported from all of the following: Streptococcus viridans, Staphylococcus epidermis, Streptococcus pneumoniae, Haemophilus, Enterococcus, Candida tropicalis and Acanthamoeba.2,4,5,13,37,38,39
Corneal granular dystrophies are a group of bilateral non-inflammatory genetic corneal diseases, the majority having no systemic association.2,3,36 Their crumb-like opacities are often mistaken for crystalline deposits.36
Progressive inherited systemic disorders may also cause the precipitation of corneal crystals, but these diseases occur earlier in life.2,3,7 Schnyder crystalline keratopathy is an autosomal-dominant, slowly progressing dystrophy that occurs in the first to second decade of life. Genetic abnormalities in lipid metabolism may cause high levels of cholesterol and phospholipid corneal deposits which increase the risk of corneal erosion.2,3,5,17,40-42
Bietti corneoretinal dystrophy is an autosomal-recessive disorder that typically occurs after the second decade of life.2,11,12,17 Presentation is characterized by bilateral crystalline deposits in the cornea and retina with progressive chorioretinal degeneration, night blindness and visual field loss.2,11,12,17 Gout is the most common inflammatory joint disease in men over 40. Monosodium urate crystals deposit in the joints and tendons from longstanding hyperuricemia.3,10 Ocular inflammation is rare but can occur during an acute attack. Uric acid crystal deposits may occur in any ocular tissue or adnexa, causing symptoms such as burning pain, redness and decreased vision.3,10,13,17 Crystal deposits may be found in the corneal epithelium, stroma and Bowman’s layer and appear as a orange-brown band keratopathy.3,10
Familial lecithin-cholesterol acyltransferase deficiency is an autosomal recessive disorder that causes unesterified cholesterol to accumulate resulting in atherosclerosis, renal insufficiency and crystalline keratopathy.3,17 Peripheral corneal arcus with stromal haze is often a concurrent finding.3,17
Cystinosis is another rare metabolic disease that causes the accumulation of the amino acid cystine within the lysosomes of cells. Crystal deposition occurs in many tissues with the kidneys and eyes the most vulnerable.
There are three clinic presentations of cystinosis based on age and severity of renal disease. Nephropathic cystinosis, the most common and severe form, occurs in infancy and leads to renal tubular Fanconi syndrome by six to 12 months of age and failure to thrive.7,8 Late-onset, or intermediate, cystinosis occurs in late childhood or early adolescence. Renal disease progression is slower with less effect on growth. Non-nephrotic cystinosis presents in adulthood with severe photophobia secondary to crystal deposition in the cornea. Other organs are not affected and kidney function is not compromised.7,8 Severe photophobia and recurrent corneal erosions are notable in this disease and often lead to the need for a corneal transplant.3,7,9
 |
Treatment options for CK vary depending on the underlying cause. In some cases, such as with MM, the condition may only be treatable and not curable. Click image to enlarge. |
Management
Multiple myeloma (MM) is not curable; it is a treatable malignancy with a median survival rate of five to eight years.21,22,25,43,45 The survival rate of MM depends on tumor stage, cytogenic abnormalities and response to therapy.21,25
Autologous stem cell transplantation (ASCT) increases median survival rate, but eligibility is based on age and comorbidities.20-22,43 The four-year survival rate for this intervention is as high as 80% for those eligible patients, with a median overall survival rate of eight years.45 In the United States, the age limit for ASCT is flexible based upon physiological rather than chronological age, so long as few comorbidities are present.20,21 Most European countries have an age limit of less than 65 years.21,43
Chemotherapy for MM is tailored by the eligibility for autologous stem cell transplantation, and survival rates have improved over the last decade. Eligibility has also increased, as the FDA has approved new chemotherapeutic agents.21,22,25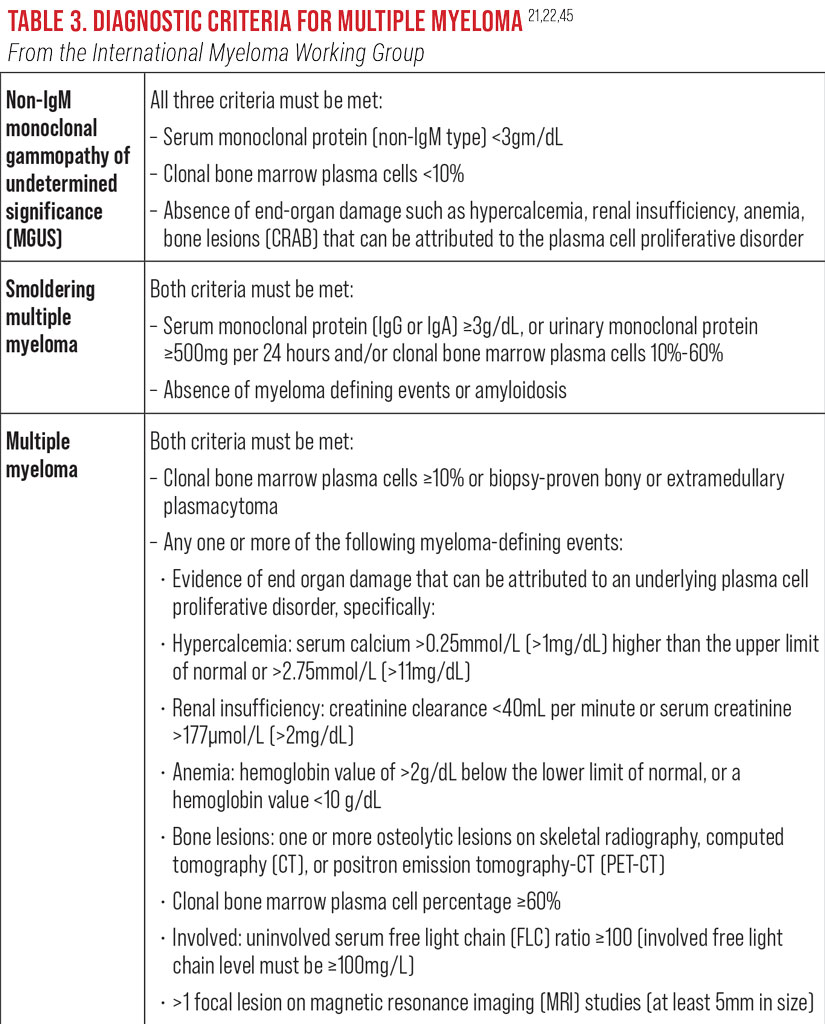
In response to chemotherapy and/or plasma exchange, a decrease in crystal density and improvement of ocular symptoms will occur as the serum immunoglobulin level decreases. Not all patients will have improvement or reduction in the corneal crystals. One study reported continued increased deposition despite systemic treatment.33-35 There are no topical ophthalmic agents that provide treatment. Topical ophthalmic agents such as artificial tears are palliative only.35,44 Confocal microscopy can be used to monitor the density of crystals as chemotherapy proceeds.33
Severely affected patients with functional vision reductions or chronic pain may require penetrating keratoplasty; unfortunately, these cases have a high risk of graft failure due to recurrence of stromal corneal opacities.35 A small number of patients may require a keratoprosthesis. Chiang et al. reported on the primary use of the Boston Type 1 keratoprosthesis, bilaterally, for a patient with severe crystalline keratopathy with vision loss.35 Visual acuity was maintained at one year of follow-up.3 Further studies using keratoprosthesis over penetrating keratoplasty are limited due to few patient numbers, low frequency of severe corneal disease and risk of complications secondary to the keratoprosthesis.35
 |
Cases of crystalline keratopathy caught early may save patients from enduring further health consequences down the line as the result of an undiagnosed systemic disease. Click image to enlarge. |
Conclusion
Crystalline keratopathy has multiple etiologies, some of which are benign, and others which are linked to potentially fatal systemic diseases. The condition itself often goes unnoticed to the patient until it progresses and produces symptoms and pain that require intervention, which can range from supportive lubricants and advanced keratoprosthesis to corneal replacement procedures. Promptly once the condition identified, a systemic workup for general illness and coagulopathy/hyperviscosity disease is warranted. Multiple myeloma is the most common of the plasma cell malignancies, as it accounts for approximately 10% of hematological malignancies, 1% of all cancers and is associated with crystalline keratopathy. It is more common in those of African descent compared with Caucasians. Although MM is not curable, early diagnosis and treatment improves quality of life and increases overall survival rate.
Any instance of crystalline keratopathy you come across in your practice calls for an immediate protocol of monitoring, testing and elimination of possible causes to identify which systemic disease, infection or other underlying issue is behind the corneal manifestation. Initiating the appropriate treatment as soon an etiology is recognized will undoubtedly improve patient outcomes. ν
Dr. Chubb is currently employed as a staff optometrist at the Michael J. Crescenz VA Medical Center in Philadelphia. She is also a fellow of the American Academy of Optometry. Dr. Chubb has no financial interests to disclose.
1. Hurley IWJ, Brooks AMV, Reinehr DP, et al. Identifying anterior segment crystals. Br J Ophthalmol. 1991;75(6):329–31. 2. Gupta PK, Kharod BV, Afshari NA, et al. Crystalline keratopathy: spectrum of disease, diagnosis and treatment. Am Acad Ophthalmol. 2007 Dec:31. www.aao.org/eyenet/article/crystalline-keratopathy-spectrum-of-disease-diagno. Accessed December 1, 2019. 3. Vinals AF, Kenyon KR. Corneal manifestations of metabolic diseases. Int Ophthalmol Clin. 1998;38(1):141–53. 4. Davison JE. Eye involvement in inherited metabolic disorders. Ther Adv Ophthalmol. 2020 Dec 29;12:2515841420979109. 5. Weiss, JS, Khemichian AJ. Differential diagnosis of schnyder corneal dystrophy. Corneal Dystrophies. Dev Ophthalmol. Basel, Karger. 2011;48:67–96. 6. Kasimer RN, Langman CB. Adult complications of nephropathic cystinosis: a systemic review. Ped Nephrol. 2021;36(2):223-36. 7. Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2(1-2):1–71. 8. Elmonem MA, Veys KR, Sokiman NA, et al. Cystinosis: a review. Orph J Rare Dis. 2016;11(47)1-17. 9. Gahl WA, Thoene JG, Schneider JA. Cystinosis. New Engl J Med. 2002;347(2):111-21. 10. Ao J, Goldblatt F, Casson RJ. Review of the ophthalmic manifestations of gout and uric acid crystal deposition. Clin and Exp Ophthalmol. 2017;45(1):73-80. 11. Manzouri B, Sergouniotis PI, Robson AG, et al. Bietti crystalline retinopathy: report of retinal crystal deposition in male adolescent siblings. Arch Ophthalmol. 2012;130(11):1470-3. 12. Song WK, Clouston P, MacLaren RE. Presence of corneal crystals confirms an unusual presentation of bietti’s retinal dystrophy. Ophthal Gen. 2019;40(5):461-5. 13. Porter AJ, Lee GA, Jun AS. Infectious crystalline keratopathy. Surv Ophthalmol. 2018;63(4): 480–99. 14. Hollander DA, Adlave AJ. Drug-Induced corneal complications. Curr Opin Ophthalmol. 2004;15(6):541-8. 15. Raizman MB, Hamrah P, Holland EJ, et al. Drug-Induced corneal epithelial changes. Surv Ophthalmol. 2017;62(3):286-301. 16. Spiegel P, Grossniklaus HE, Reinhart WJ, et al. Unusual presentation of paraproteinemic corneal infiltrates. Cornea. 1990;9(1):81-5. 17. Mazzotta C, Caragiuli S, Caporossi A. Confocal microscopy in a case of crystalline keratopathy in a patient with smouldering multiple myeloma. Int Ophthalmol. 2014;34(3):651-4. 18. Choulakian MY. Hematologic diseases and malignancies. In: Mannis MJ, Holland EJ eds. Cornea. 4th ed. Elsevier. 2017:688–95. 19. Knapp AJ, Gartner S, Henkind P. Multiple myeloma and its ocular manifestations. Surv Ophthalmol. 1987;31(5):343–51. 20. Michels TC, Petersen KE. Multiple myeloma: diagnosis and treatment. Am Fam Physician. 2017;95(6):373-85. 21. Rajkumar SV, Kumar S. Multiple Myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91(1):101–19. 22. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93(8);1091-1110. 23. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a population-based study. Blood. 2010;116(25);5501-6. 24. Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev. 2016;30(3):223-31. 25. Panopoulou A, Streetly MJ. Myeloma and MGUS. Med. 2017;5(45):311-17. 26. Koo H, Oh D, Chun YS, et al. A case of crystalline keratopathy in monoclonal gammopathy of undetermined significance (MGUS). Korean J Ophthalmol. 2011;25(3):202-5. 27. Karakus S, Gottsch JD, Caturegli P, et al. Monoclonal gammopathy of ‘ocular’ significance. Am J Ophthalmol Case Rep. 2019 May 20;15:100471. 28. Kunacheewa C, Manasanch EE. High-risk smoldering myeloma versus early detection of multiple myeloma: current models, goals of therapy, and clinical implications. Best Prac and Res Clin Haematol. 2020;33(1):101152. 29. Orellana J, Friedman AH. Ocular manifestations of multiple myeloma, waldenström’s macroglobulinemia and benign monoclonal gammopathy. Surv Ophthalmol. 1981;26(3):157-69. 30. Fung S, Selva D, Leibovitch I, et al. Ophthalmic manifestations of multiple myeloma. Ophthalmologica. 2005;219(1):43–8. 31. Balderman SR, Lichtman MA. Unusual manifestations of monoclonal gammopathy: ocular disease. Rambam Maimonides Med J. 2015;6(3):1-15. 32. Henderson DW, Stirling JW, Lipsett J, et al. Paraproteinemic crystalline keratopathy: an ultrastructural study of two cases, including immunoelectron microscopy. Ultrastructural Path. 1993;17(6):643-68. 33. Buerk BM, Tu E. Confocal microscopy in multiple myeloma crystalline keratopathy. Cornea. 2002;21(6):619-20. 34. Garibaldi DC, Gottsch J, De La Cruz Z, et al. Immunotactoid keratopathy: a clinicopathologic case report and a review of reports of corneal involvement in systemic paraproteinemias. Surv Ophthalmol. 2005;50(11):61-79. 35. Chiang H, Wieland RS, Rogers TS, et al. Paraproteinemic keratopathy in monoclonal gammopathy of undetermined significance treated with primary keratoprosthesis. Medicine (Baltimore). 2017;96(50):50-6. 36. Klintworth, GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4(7):1-38. 37. Karani R, Sherman S, Trief D. A case of infectious crystalline keratopathy after cross-linking. Am J Ophthalmol Case Rep. 2021;23:101139. 38. Modabber M, Darvish-Zargar M, Breton, L, et al. Crystalline keratopathy in post-lasik ectasia: a case report. Cornea. 2019 May;38:635-8. 39. James CB, McDonnell PJ, Falcon MG. Infectious crystalline keratopathy. Br J Ophthalmol. 1988;72(8):628-30. 40. Karseras AG, Price DC. Central crystalline corneal dystrophy. Brit J Ophthalmol. 1970; 54(10):659-61. 41. Bron AJ, Williams HP, Carruthers ME. Hereditary crystalline stromal dystrophy of schnyder. Brit J Ophthalmol. 1972;56(5):383-99. 42. Burns RP, Connor W, Gipson I. Cholesterol turnover in hereditary crystalline corneal dystrophy of schnyder. Trans Am Ophthalmol Soc. 1978;76:184-96. 43. Fazel F, Bassa F. An approach to the diagnosis and management of multiple myeloma. S Afr Med J. 2019;109(10):723-27. 44. Kleta R, Blair SC, Bernardini I, et al. Keratopathy of multiple myeloma masquerading as corneal crystals of ocular cystinosis. Mayo Clin Proc. 2004;79:410-12. 45. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(5):548-67. |
