A fundamental understanding of fetal ocular development can greatly increase your knowledge of these anomalous conditions, and will help guide your diagnostic testing and management protocol. Here, we’ll review the signs and symptoms of the most common congenital anomalies you’ll encounter in clinical practice.
|
Exploring the Eye in Utero
During the fourth week of embryonic gestation, two optic pits begin to form. These bumps proliferate to form hollow vesicles, which slowly adhere to the developing spinal column to form optic stalks. As the cellular contents of the optic vesicles increase, their proximity to the adjacent surface ectoderm induces tissue invagination. This creates both the optic cup and a lens placode. Asymmetric invagination and unequal cellular proliferation rates produce a groove that evolves into the optic or fetal fissure. This inferior opening closes by the seventh week of fetal development.1,2 Neuroectoderm cells, positioned apex to apex within the inner layer of the optic cup, form the neurosensory retina––with the outer cell layer forming the retinal pigment epithelium (RPE). Next, upon further invagination, the optic nerve forms from the optic stalks. This also creates a bilayer architecture, which forms the optic nerve and its surrounding sheath. Myelination occurs during the last four months of gestation. Once the optic nerve fibers have reached the lateral geniculate nucleus (LGN), myelination begins and proceeds from the LGN to the chiasm, eventually terminating at the lamina cribrosa near birth.3 Many of the remaining ocular structures develop over time as the tissues mature. Waves of mesenchyme (a tissue chiefly comprised of mesoderm and neural crest cells) produce the sclera, which arises from limbal tissue and projects back until it reaches the optic nerve. The complex network of choroidal vessels develops from mesenchyme proximal to the future RPE.2 With respect to ocular vasculature, the central retinal artery begins as a small vessel that penetrates the fetal groove prior to its closure. In its earliest stages, the vessel forms the fetal hyaloid artery, which branches in the vitreous cavity to form a network of vessels known as the posterior tunica vasculosa lentis. During this time, glial cells located adjacent to the optic cup form a sheath that surrounds the base of the hyaloid artery.2 Under normal circumstances, the hyaloid system degenerates––ensuring that the vascular network is completely reabsorbed before birth. Simultaneously, as the hyaloid complex develops and atrophies, the permanent retinal vascular system develops, sending early branches into the retinal nerve fiber layer.2 The entire vascular bed develops from month four of gestation to month three postpartum.2 |
Anophthalmos is defined as complete agenesis of the globe. The condition is caused by optic vesicle atrophy very early in embryogenesis.1,4,5
Microphthalmos is characterized by an abnormally small globe and correspondingly small orbit. A microphthalmic eye is the result of arrested tissue development with partial differentiation.1,6
Both conditions are widely believed to be induced by genetic defects, and are leading causes of congenital blindness.1,4,5
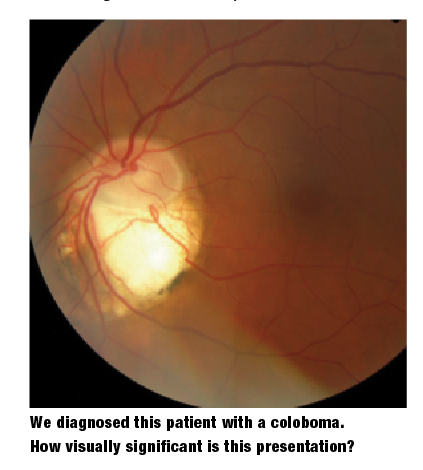
Coloboma
Posterior colobomas develop secondary to incomplete optic fissure closure, and may occur at any position along its length. As the tissue fails to close, the developing fetus experiences overlying retinal, choroidal and optic nerve hypoplasia.
A coloboma of the optic disc presents as an excavation of the inferior rim tissue, and is often accompanied by abnormal glial cells.1,7,8 A retinal coloboma, on the other hand, often manifests with the characteristic bare sclera appearance, and usually is accompanied by hypoplasia or aplasia of the overlying choroidal and retinal tissue.1,7,8
A posterior coloboma’s location and severity dictates its functional significance. Given proper development of the fovea and optic disc, retinal colobomas rarely affect acuity.9 Secondary clinical sequelae from concurrent peripapillary changes can cause serous maculopathy (central serous chorioretinopathy). In such instances, vision acutely changes as the neurosensory retina detaches from the RPE.1,8
An optic nerve coloboma can mimic glaucomatous cupping. Anomalous retinal vasculature exiting the optic disc can aid in discriminating an optic disc coloboma from glaucomatous cupping.8 Interestingly, the literature indicates that discs with colobomatous signs have increased fragility, even in the context of non-hypertensive intraocular pressure.1,8 This is an important consideration, because clinicians should photodocument and closely monitor these anomalies for pathological conversion.1,8
Tilted Disc Syndrome
This condition is believed to result from either incomplete optic fissure closure or sectoral hypoplasia.10 Tilted disc syndrome (TDS) has a variable presentation with a range of physical appearances.
The general perception that TDS induces inferior or inferonasal disc tilting, with some degree of rotation along the vertical axis, is not precisely accurate.10,11 Instead, incomplete disc formation causes the appearance of ectasia accompanied by varying degrees of hypoplasia in the choroid, retina and RPE––yielding the appearance of rotation.10,11
These changes increase the inherent risk of choroidal neovascularization secondary to cytokine release from morphological tissue abnormalities.10
A reversed vascular branching pattern known as “situs inversus” often accompanies TDS. This presentation is characterized by nasal deviation of the retinal vasculature upon exiting the disc that includes a sudden temporal turn to form the major arcades. The anomalous pattern likely is due to atypical morphology of the scleral canal, through which the optic nerve and its accompanying vasculature traverse.12 Although benign, the pattern may make optic disc evaluation more challenging.10,13 The idea that individuals who exhibit situs inversus have systemic organ dislocation is a myth.10-13
 Patients with TDS may exhibit clinical signs that mimic those associated with acquired optic disc pathologies, such as glaucomatous cupping.10 Adding to the conundrum, atypical nerve fiber distribution and segmental underdevelopment can produce depressed visual fields or various scotomata. Because the corresponding field defects often are located superotemporally, clinicians may erroneously diagnose the condition as neurological loss or a budding glaucomatous pathology.10,11 Unlike patients with the aforementioned conditions, however, those with TDS may experience fluctuating visual field results via perimetry testing.10,11
Patients with TDS may exhibit clinical signs that mimic those associated with acquired optic disc pathologies, such as glaucomatous cupping.10 Adding to the conundrum, atypical nerve fiber distribution and segmental underdevelopment can produce depressed visual fields or various scotomata. Because the corresponding field defects often are located superotemporally, clinicians may erroneously diagnose the condition as neurological loss or a budding glaucomatous pathology.10,11 Unlike patients with the aforementioned conditions, however, those with TDS may experience fluctuating visual field results via perimetry testing.10,11
Optic Pit
Optic pits are single, round or oval depressions in the temporal or inferotemporal aspect of the optic cup.14,15 They may be observed exiting the choroidal or retinal vessels.14,15 Some pits are veiled by a sheet of glial tissue, making them more difficult to discern.15 Optic pits vary in size, depth and coloration, and tend to present unilaterally.14
Some researchers consider optic pits to be atypical colobomatous defects of the nerve itself––although the pathogenesis is unclear.16,17 Histologically, optic pits are invaginations of the optic disc that contain neuroretinal tissues.14,15 Acuity loss is common when optic pits are present due to corresponding macular pathologies.
Central serous chorioretinopathy (CSCR), cystic macular edema, macular holes and macular cysts frequently are associated with optic pits.15,16,18 The origin of fluid leakage in CSCR is unclear, but is hypothesized to originate from either liquefied vitreous humor trapped within the pit or cerebrospinal fluid leakage from the pit to the subarachnoid space.15,16 An atypical vitreous attachment at the optic disc and/or an acquired break in the thin retinal tissue overlying or adjacent to the optic pit likely enable fluid passage (wicking), producing a shallow, rhegmatogenous neurosensory retinal detachment.23
Optic pits are often mistakenly identified as acquired optic neuropathy because of their characteristic nerve coloration changes (relative pallor), or as glaucomatous cupping. Optic pits have the potential to affect visual acuity depending on the tissues involved, and frequently produce deep, fluctuating visual field defects.15-18
 |
|
Optic disc drusen, as seen here, may be challenging to differentiate from disc edema.
|
Optic Disc Hypoplasia
Optic disc hypoplasia is the result of optic nerve underdevelopment during gestation.19-21 The etiology is unclear, and environmental causes (maternal drug, alcohol and tobacco use), circumstances of pregnancy (viral infection, prematurity or first child) and gene expression all have been implicated.20
Hypoplastic discs have a small diameter (<1,500µm), pale or gray coloration, and are surrounded by the characteristic “double ring sign”––which is formed via retinal tissue extension over the lamina cribrosa and scleral canal.20,22 This telltale clinical sign manifests when the physically smaller disc is placed within a normally-sized scleral canal.
Depending on the quantity of axons in the papillomacular nerve fiber bundle, visual acuity may range from normal to no light perception. Optic nerve hypoplasia is recognized as a common cause of congenital blindness.19,20,23 Visual field defects, strabismus (when monocular or asymmetric hypoplasia) and nystagmus may accompany the reduced function.21 Fortunately, any areas of functional vision generally remain stable over time.
Optic disc hypoplasia may exist in concert with midline defects, such as septum pellucidum and/or corpus callosum agenesis.19,20,23 Hypopituitarism (due to hypothalamus dysfunction), hypothyroidism and decreased growth hormone are possible endocrine comorbidities.19-23
Morning Glory Syndrome
This disc anomaly typically presents unilaterally, and is characterized by excavation.13,19 The condition also produces an atypical vascular appearance. Blood vessels appear to exit from the peripheral disc in a radial pattern.13,19
Due to bifurcations at the optic disc, the total number of blood vessels appears to be more numerous than in a normal individual.13,19 Because a white glial tuft often is present centrally, the exiting vasculature is further skewed. A circumferential annulus of pigment disruption characteristically surrounds the disc.
The etiology of morning glory syndrome is uncertain. Some have proposed that the associated disc anomaly is located along the coloboma’s continuum, while others suggest that it is a completely separate and sporadic dysplasia.13,19 While vision may range from normal to hand motion, patients with morning glory syndrome may develop associated vision loss if nonrhegmatogenous retinal detachments occur.13,19
Myelinated Nerve Fibers
In approximately 1% of individuals, myelination extends beyond the lamina cribrosa and into the retina, appearing as a visible element of the nerve fiber layer.3,13 Myelination of the retinal ganglion cell axons generates a white and feathery obstruction within the NFL that may or may not abut the optic nerve head.
Single isolated patches, arcuate-shaped regions or multiple separated patches (usually in the posterior pole) also may exist. Due to their opaque density, however, underlying blood vessels cannot be seen upon clinical evaluation.
Myelination may create the appearance of leukocoria (white coloration that’s visible inside the pupil). Additionally, the condition must be differentiated from cotton- wool spots––which exhibit a white, fluffy appearance produced by retinal ganglion cell axonal death with associated axoplasmic stasis (slowed cellular conduction along the nerve).
Because myelination has the ability to block light from reaching the photoreceptors, its presence may create relative scotomata upon visual field testing.13 Although myelinated nerve fibers are a benign congenital finding, affected individuals are at an increased risk for regression in neurodegenerative conditions (e.g., glaucoma).3
Peripapillary Crescents
Scleral and choroidal crescents typically occur temporal to the optic disc; they are benign anatomical variations. A scleral crescent results when retinal and choroidal tissue fail to directly abut the optic nerve head (ONH), which permits direct visualization of the sclera.13 Similarly, choroidal crescents result from the absence of retinal tissue directly adjacent to the ONH, allowing more detailed visualization of the underlying choroidal pigment in the region.
Crescents may change over time with globe elongation or other pathologies that cause retinal stretching.13 Choroidal crescent pigment may mimic acquired peripapillary atrophy secondary to an optic neuropathy. These crescents also may enlarge the blind spot measurement during perimetry.13
Incomplete Regression of the Hyaloid Vasculature
Incomplete atrophy and regression of the fetal hyaloid system can result in either benign or pathological retention of the infantile blood vessel system. Bergmeister’s papilla is an example of benign glial remnants positioned on or over the optic nerve head.13 They are pathologic when any original hyaloid vessels remain patent. (It is important to note that patent hyaloid vessels may lead to spontaneous vitreous hemorrhage.25,26)
A more serious condition occurs when hyaloid regression is unsuccessful, provoking a condition known as persistent hyperplastic primary vitreous (PHPV).25,26 Here, the abnormally retained fetal vascular system induces fibrovascular mass growth. The potential for a fibrovascular mass to cause further pathology is profound, and includes premature cataractogenesis, retinal detachment, microphthalmos and angle-closure glaucoma.26
PHPV must be differentiated from both retinal detachment and malignant mass growth (retinoblastoma). Differentiation may be assisted by Doppler imaging, which provides a 2D rendering of the lesion.25
This condition is rare in infants who were carried to full term. However, some degree of fetal vasculature persistence is common in infants who are born prematurely.
 |
| This patient shows evidence of nerve fiber myelination. |
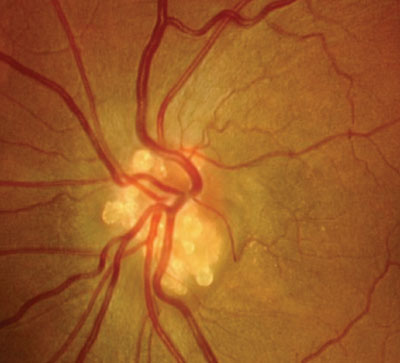
Optic disc drusen are accumulations of acellular calcified globular material deposited anterior to the lamina cribrosa in the optic nerve head.13,27,28 Disc drusen occur in approximately 1% of the population.13 They typically develop in whites and are inherited in an irregular autosomal dominant pattern.13 Also, optic disc drusen often present bilaterally.13
In the earliest years of life, drusen tend to be buried. With time they migrate anteriorly, becoming more visible at the level of the optic disc rim––often creating a lumpy, refractile appearance.13
The presence of disc drusen can cause both structural and functional deficits. For instance, buried optic disc drusen can move to superficial locations in the optic disc, compressing optic nerve blood vessels and ganglion cell axons. The compression can induce both vein and central retinal artery occlusion, as well as alter axoplasmic flow. In such instances, the compression can kill affected cells, resulting in functional vision loss and nerve fiber dropout.13
Because the position of optic disc drusen can vary throughout life, associated visual field deficits may slowly change or progress. Optic disc drusen density and sharpness also may cause nicking or shearing of peripapillary blood vessels. This can produce intraretinal disc hemorrhages.13
Optic disc drusen can pose a diagnostic dilemma, because the presentation may resemble disc edema (pseudopapilledema) or may even be accompanied by true disc edema.13,28 Suspicion of disc edema warrants a comprehensive diagnostic evaluation.
Failure to differentiate optic disc drusen from genuine disc edema may subject the patient to invasive, unsubstantiated neurological testing. B-scan ultrasonography, optical coherence tomography, computed tomography, fluorescein angiography and fundus autofluorescence imaging are useful for the detection of disc drusen.25,28 Other, less common differentials may include astrocytic hamartoma and optic nerve meningiomas.2
In most instances, congenital anomalies of the optic disc remain stable throughout life, have little effect on visual function and require no treatment. However, in some instances––based on their developmental etiology––associated ocular and systemic signs can produce changes that precipitate secondary pathologies that can affect the patient’s vision.
Understanding the etiologies of these commonly observed findings will enable you to properly educate affected individuals and prescribe appropriate testing, documentation and monitoring for potential complications.
Dr. Hutchinson McGinnis practices in St. Louis and is an assistant clinical professor at the University of Missouri-St. Louis College of Optometry.
Dr. Gurwood is a professor at Salus University in Elkins Park, Pa.
1. Oyster CW. Formation of the human eye. In: Oyster CW. The Human Eye: Structure and Function. Sunderland, Mass.: Sinauer Associates, Inc; 1999:57-72.
2. Remington LA. Ocular embryology. In: Clinical Anatomy and Physiology of the Visual System. St Louis: Elsevier; 2012:123-43.
3. Sowka JW, Nadeau MJ. Regression of myelinated retinal nerve fibers in a glaucomatous eye. Optom Vis Sci. 2013;90(7):e218-20.
4. Skalicky SE, White AJ, Grigg JR, et al. Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum. JAMA Ophthalmol. 2013 Dec;131(12):1517-24.
5. Chassaing N, Causse A, Vigouroux A, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin Genet. 2013 Sep 10. doi: 10.1111/cge.12275. [Epub ahead of print]
6. Duke-Elder S. Mircophthalmos. In: Duke-Elder S (ed.). System of ophthalmology, Vol III. Normal and abnormal development, part 2. Congenital Deformities. London: Henry Kimpton;1964:488-95.
7. Lin CC, Tso MO, Vygantas CM. Coloboma of optic nerve associated with serous maculopathy: a clinicopathologic correlative study. Arch Ophthalmol. 1984 Nov;102(11):1651-954.
8. Savell J, Cook JR. Optic nerve colobomas of autosomal-dominant heredity. Arch Ophthalmol. 1976 Mar;94(3):395-400.
9. Olsen TW, Summers CG, Knobloch WH. Predicting visual acuity in children with colobomas involving the optic nerve. J Pediatr Ophthalmol Strabismus. 1996 Jan-Feb;33(1):47-51.
10. Sowka J, Aoun P. Tilted disc syndrome. Optom Vis Sci.1999;76(9):618-23.
11. Vuori ML, Mäntyjärvi M. Tilted disc syndrome may mimic false visual field deterioration. Acta Ophthalmol. 2008 Sep;86(6):622-5.
12. Young SE, Walsh FB, Knox DL. The tilted disk syndrome. Am J Ophthamol. 1976 Jan;82(1):16-23.
13. Gaddie IB, Alexander LJ. Congenital and acquired anomalies of the optic nerve head. In: Alexander LJ (ed.). Primary Care of the Posterior Segment. New York: McGraw-Hill; 2002:209-325.
14. Kranenburg EW. Crater-like holes in the optic disc and central serous retinopathy. Arch Ophthalmol. 1960 Jun;64(6):912-24.
15. Corbett JJ, Savino PJ, Schatz NJ, et al. Cavitary developmental defects of the optic disc. Arch Neurol. 1980 Apr;37(4):210-3.
16. Taylor D. Developmental abnormalities of the optic nerve and chiasm. Eye (Lond). 2007 Oct;21(10):1271-84.
17. Gregory-Roberts EM, Mateo C, CorcÓstegul, et al. Optic disk pit morphology and retinal detachment: optic coherence tomography with intraoperative correlation. Retina. 2013 Feb;33(2):363-70.
18. Gass JD. Serous detachment of the macula. Secondary to congenital pit of the optic nervehead. Am J Ophthalmol. 1969 Jun;67(6):621-41.
19. Brodsky MC. Congenital optic disc anomalies. In: Yanoff M, Duker JS. Ophthalmology. Philadelphia: Mosby; 2009:956-9.
20. Wall PB, Traboulsi EI. Congenital abnormalities of the optic nerve: from gene mutation to clinical expressions. Curr Neurol Neurosci Rep. 2013 Jul;13(7):363.
21. Brodsky MC, Glasier CM, Pollock SC, et al. Optic nerve hypoplasia. Identification by magnetic resonance imaging. Arch Ophthalmol. 1990 Nov;108(11):1562-7.
22. Borchert M. Reappraisal of the optic nerve hypoplasia syndrome. J Neuroophthalmol. 2012 Mar;32(1):58-67.
23. Postel EA, Pulido JS, McNamara JA, et al. The etiology and treatment of macular detachment associated with optic nerve pits and related anomalies. Trans Am Ophthalmol Soc. 1998;96:73-88.
24. Blinder K. Personal communication: Dec 2013.
25. Neudorfer M, Waisbourd M, Buzi S, et al. Color Doppler imaging of eyes with persistent fetal vasculature. Pediatr Radiol. 2012 Oct;42(10):1229-34.
26. Townsend W, Alexander LJ. Anomalies of the vitreous and peripheral retina. In: Alexander LJ. Primary Care of the Posterior Segment. New York: McGraw-Hill; 2002:471.
27. Gili P, Flores-Rodriguez P, Martin-Rios MD, et al. Anatomical and functional impairment of the nerve fiber layer in patients with optic nerve head drusen. Graefes Arch Clin Exp Ophthalmol. 2013 Oct;251(10):2421-8.
28. Sarac O, Tasci YY, Gurdal C, Can I. Differentiation of optic disc edema from optic nerve head drusen with spectral-domain optical coherence tomography. J Neuro-ophthalmol. 2012 Sep;32(3):207-11.
