 |
A 17-year-old Chinese male presented for evaluation of a presumed inherited retinal dystrophy. He had a longstanding history of poor night vision and carried a clinical diagnosis of retinitis pigmentosa (RP) since age six. The patient had two younger brothers, who shared the same biological parents and symptom of poor night vision. The parents denied consanguinity.
His best-corrected visual acuities were 20/40 OD and 20/50 OS. His intraocular pressures were 11mm Hg OD and 12mm Hg OS, extraocular motilities were full, confrontation visual fields were mildly constricted in both eyes, and pupils were equally round and reactive with no relative afferent pupillary defect. Anterior segment exam was unremarkable OU. Posterior segments contained bone spicule-like pigmentary changes in a midperipheral annular configuration and macular pigment changes. The optic nerves had an atypical presentation for which various studies are presented herein (Figures 1-4). The retina was otherwise flat and attached in both eyes with no other observed lesions OU.
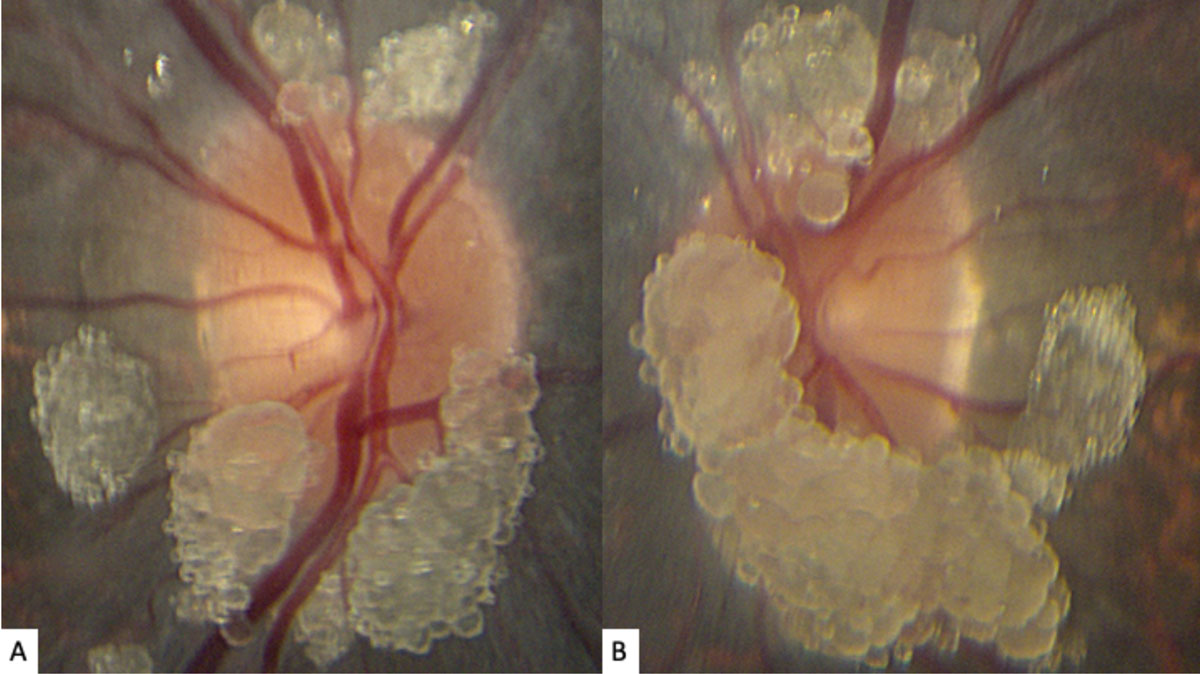 |
| Fig. 1. These photos of our young patient’s right (A) and left (B) optic nerves show extensive elevated, globular lesions of both the optic nerve and peripapillary retina. Click image to enlarge. |
Take the Retina Quiz
1. Which of the following best describes the fundus autofluorescence findings depicted in Figure 2?
a. Hyperfluorescence of lesions at the optic nerve.
b. Hyperautofluorescence of the lesions at the optic nerve.
c. Hypofluorescence of the lesions at the optic nerve.
d. Hypoautofluorescence of the lesions at the optic nerve.
2. Optical coherence tomography (OCT) imaging suggests that this lesion arises from which ocular structure (Figures 3 and 4)?
a. The vitreous.
b. The inner retina.
c. The outer retina.
d. The retinal pigment epithelium.
3. Which of the following is the most likely diagnosis of the optic nerve finding?
a. Retinoblastoma.
b. Optic nerve head drusen.
c. Retinal/optic nerve astrocytic hamartoma.
d. Retinal hemangioblastoma.
4. This lesion has been associated with which of the following conditions:
a. Tuberous sclerosis complex.
b. Neurofibromatosis.
c. Retinitis pigmentosa.
d. The lesions are associated with all of the above conditions.
5. Which of the following statements regarding the lesion is false?
a. They are composed of glial cells.
b. They are relatively benign, slow-growing tumors.
c. They are composed of hyaline bodies.
d. They can cause exudative retinal detachments.
For answers, see below.
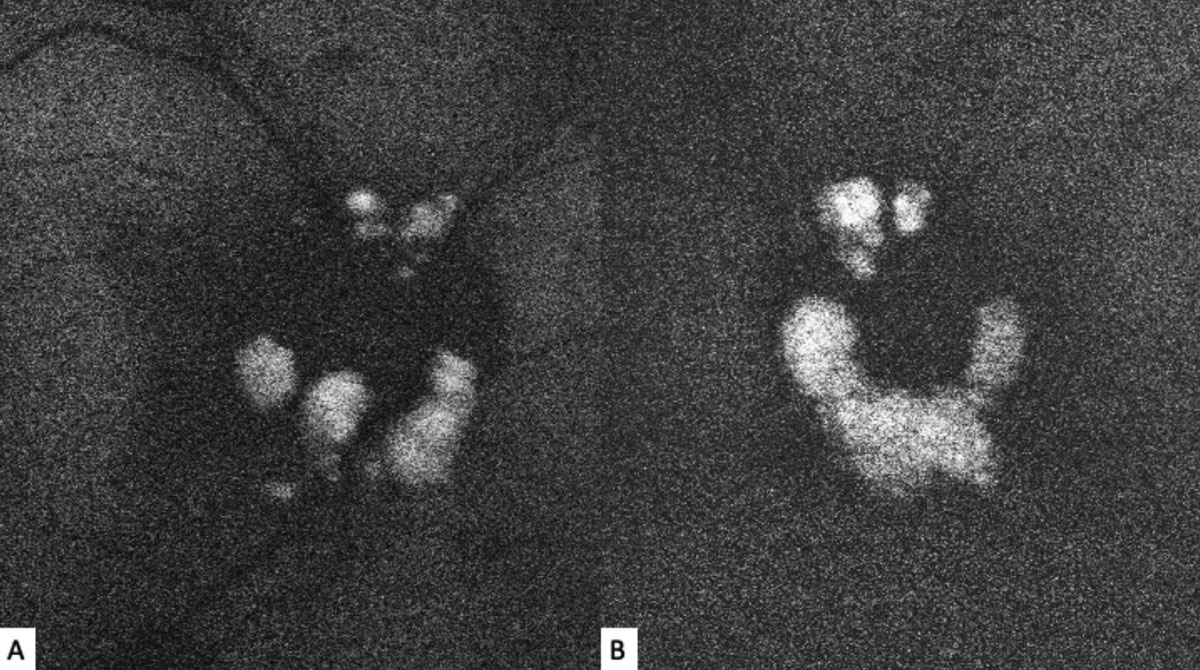 |
| Fig. 2. Fundus autofluorescence of the right (A) and left (B) optic nerves show discrete hyperautofluorescence corresponding with the lesions seen in Figure 1. Click image to enlarge. |
Diagnosis
The striking appearance of both optic nerves show mulberry-like lesions of the optic nerve and peripapillary retina in both eyes (Figures 1a and 1b). Even though the lesions were elevated, the optic disc margins were sharp and the nerve itself was flat with no presence of fluid. Fundus autofluorescence (FAF) imaging showed hyperautofluorescence of the lesions in both eyes (Figures 2a and 2b). OCT imaging revealed hyporeflective optically empty spaces of the inner retina that appear confined to the retinal nerve fiber layer (RNFL) with focal intralesional hyper-reflective opacities (Figures 3 and 4). The lesions were clinically consistent with astrocytic hamartomas of the retina and optic nerve.
Discussion
Retinal and optic nerve astrocytic hamartomas (RAH) are relatively rare, benign tumors of the retina and optic nerve that can be unilateral or bilateral.1,2 They are most commonly seen in tuberous sclerosis complex (TSC), a systemic disorder defined by the triad of epilepsy, mental retardation and skin lesions that typically affect the head and face. In fact, RAHs are the most common ophthalmic manifestation of TSC and are present in up to 53% of cases.1,3-6 In addition to TSC, RAHs have also been described in neurofibromatosis, RP or in isolation.3,4
Clinically, they appear as elevated, globular, yellow-white or translucent nodules; their appearance has been compared to that of fish eggs, tapioca and mulberries.1,2,5,6,9,10 On OCT, they are RNFL tumors with hyporeflective, “moth-eaten” optically empty spaces with foci of hyperreflectivity, which researchers believe represent intralesional calcifications.5,10-12 When calcified, the lesions hyperautofluoresce on FAF similar to optic nerve head drusen (ONHD).5 While such findings are thought to be avascular, investigators have described cases where mild vascularity is evidenced on fluorescein angiography.8 Vascularity can result in exudation and subsequent vision loss by way of retinal detachment.8
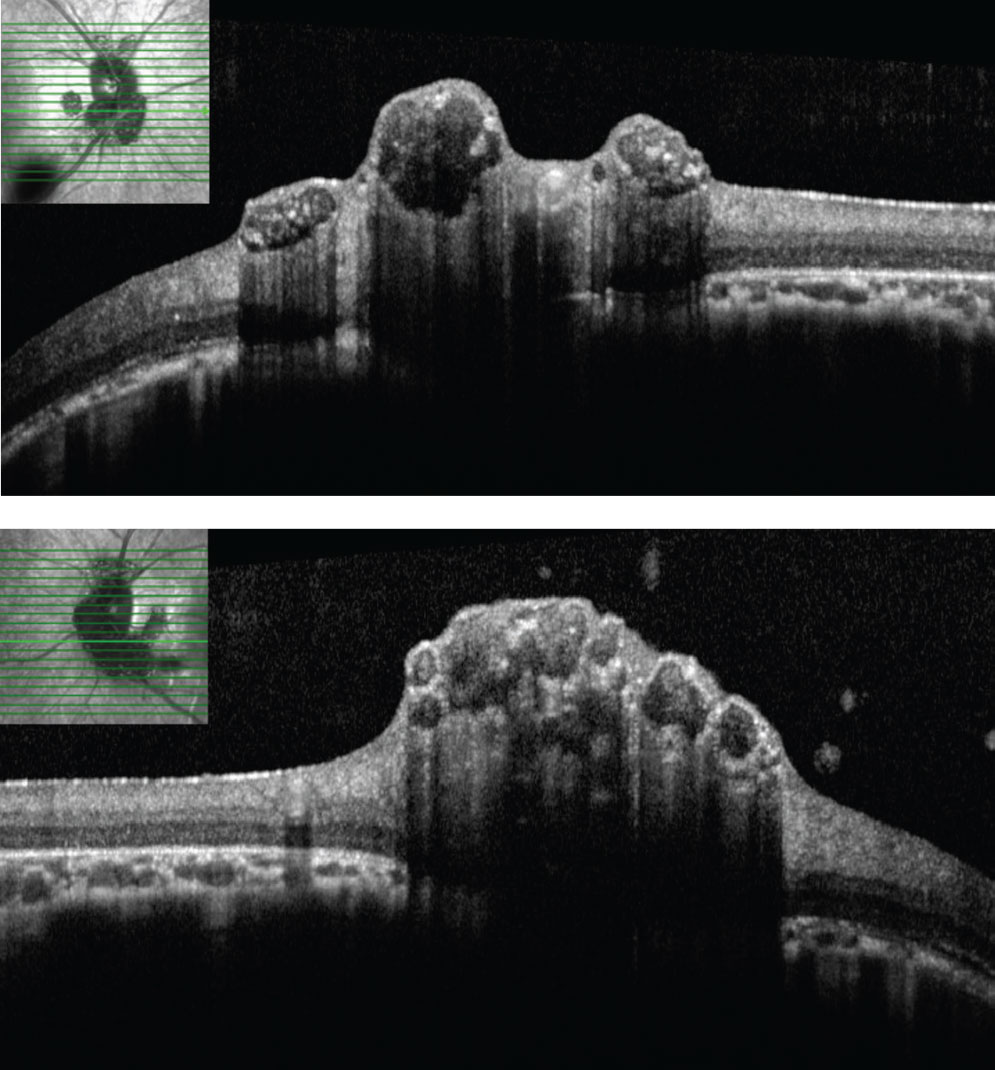 |
| Figs. 3 and 4. SD-OCT through lesions of the right optic nerve. On SD-OCT, the lesions appear to have hypo-reflective “moth eaten,” optically empty spaces with foci of hyperreflectivity, likely reflecting areas of intralesional calcification. Click image to enlarge. |
Typically, RAHs are endophytic tumors (arising from the RNFL and bulging toward the vitreous), but can rarely be exophytic (arising from the subretinal space).1,5,6 Exophytic tumors tend to be more visually significant as a result of exudative retinal detachments and subsequent neovascular glaucoma.1,6 Histologically, RAHS are RNFL tumors composed of fibrillary astrocytes (glial cells); they typically begin as flat lesions that slowly progress to elevated nodules in the first few decades of life with variable foci of calcification.1,3,5,7-10
Differential diagnoses include retinoblastoma, ONHD, acquired retinal astrocytoma, retinal hemangioblastoma, posterior amelanotic uveal melanoma, choroidal metastases and Coats’ disease. Cavitary retinoblastoma, a unique variant of retinoblastoma of low-grade malignancy, is characterized by similar OCT features of focal hyperreflective opacities within hyporeflective cavities, making distinction especially challenging.10,13 In contrast to RAHs, ONHD are hyaline bodies within the substance of the optic nerve that undergo calcification with age.7,8 By virtue of anatomy, ONHD result in a congested or elevated appearance to the optic nerve, while RAHs rest superficial to a flat optic nerve with distinct margins.7,8
Management of RAHs involves observation.1,5,8 While ophthalmic complications are rare, they include retinal neovascularization, vitreous hemorrhage or seeding, macular edema, intraretinal or subretinal exudation and neovascular glaucoma.1,5,8
Treatment options include intravitreal anti-VEGF, intravitreal steroids, photodynamic therapy and pars plana vitrectomy either in combination or isolation.1,5,8
Our patient indeed had RP, in addition to the RAH. Given that both parents were asymptomatic and that all three male kin exhibited the disease, it was presumed to be of autosomal recessive or X-linked recessive inheritance. A blood sample was obtained and sent for genetic analysis. Genetic testing revealed a mutation in the RPGR gene, which is known to be pathogenic for X-linked RP. A genetically confirmed diagnosis of X-linked RP was obtained, and the patient and family were genetically counseled.
Although our patient did not have any apparent systemic manifestations of TSC, he was recommended to follow-up with internal medicine for further studies to rule it out.
Dr. Aboumourad practices at the Bascom Palmer Eye Institute in Miami.
Retina Quiz Answers
1) b; 2) b; 3) c; 4) d; 5) c.
1. Ryan S. Retina. 4th ed. Vol I-III. Philadelphia: Mosby Elsevier; 2006. 2. Yanoff M, Duker JS, eds. Ophthalmology. 5th ed. Philadelphia: Elsevier; 2019. 3. Saxena S, Meyer C. Peripapillary astrocytic hamartomas evolve from the optic nerve. BMJ Case Rep. 2015; 2015. 4. Kinori M, Moroz I, Rotenstreich Y, et al. Bilateral presumed astrocytic hamartomas in a patient with retinitis pigmentosa. Clin Ophthalmol. 2011;5:1663-5. 5. Taylor D, Hoyt CS, eds. Pediatric Ophthalmology and Strabismus. New York: Elsevier Saunders; 2013. 6. Bagheri N, Wajda B, Calvo C. The Wills Eye Manual: Office and Emergency Room Diagnosis and Treatment of Eye Disease. 7th Ed. Philadelphia: Lippincott Williams & Wilkins; 2016. 7. Shields J. Tumors and pseudotumors of the optic disc. Acta Ophthalmol Scand. 2000;78(2):156-63. 8. Gass J. Stereoscopic atlas of macular disease : a fundoscopic and angiographic presentation. Saint Louis: Mosby; 1970. 9. Freund K, Sarraf D, Mieler W, Yannuzzi L. The retinal atlas. 2nd ed. New York: Saunders-Elsevier; 2017. 10. Shields C, Manalac J, Das C, et al. Review of spectral domain-enhanced depth imaging optical coherence tomography of tumors of the retina and retinal pigment epithelium in children and adults. Ind J Ophthalmol. 2015;63(2):128-32. 11. Shields C, Benevides R, Materin M, Shields J. Optical coherence tomography of retinal astrocytic hamartoma in 15 cases. Ophthalmol. 2006;113(9):1553-7. 12. Shields C, Say E, Fuller T, et al. Retinal astrocytic hamartoma arises in nerve fiber layer and shows “moth-eaten” optically empty spaces on optical coherence tomography. Ophthalmol. 2016;123(8):1809-16. 13. Fuller T, Alvi R, Shields C. Optical coherence tomography of cavitary retinoblastoma. JAMA Ophthalmol. 2016;134(5):e155355. |
