 |
A 34-year-old Caucasian male presented to the clinic with complaints of progressive blurry central vision he has experienced since childhood. He’s also experienced trouble seeing at night. The patient reported a male cousin and maternal grandfather suffering similar vision loss, which progressed later in life.
On examination, the patient’s best-corrected visual acuities were 20/40 OD, 20/60 OS with a prescription of -7.00D OD, -6.75D OS. Pupils were sluggish, with minimal response to light, confrontation visual fields were constricted to a central 30 degrees in each eye and extraocular muscles were full in the presence of horizontal nystagmus in both eyes.
Color vision, measured with D-100 color plate, showed only eight correct with the right eye, and seven with the left. Intraocular pressures (IOPs) were 16mm Hg OD and 14mm Hg OS with Tonopen (Reichert). Dilated fundus exams were also performed (Figure 1). Additionally, we obtained OCT images (Figure 2).
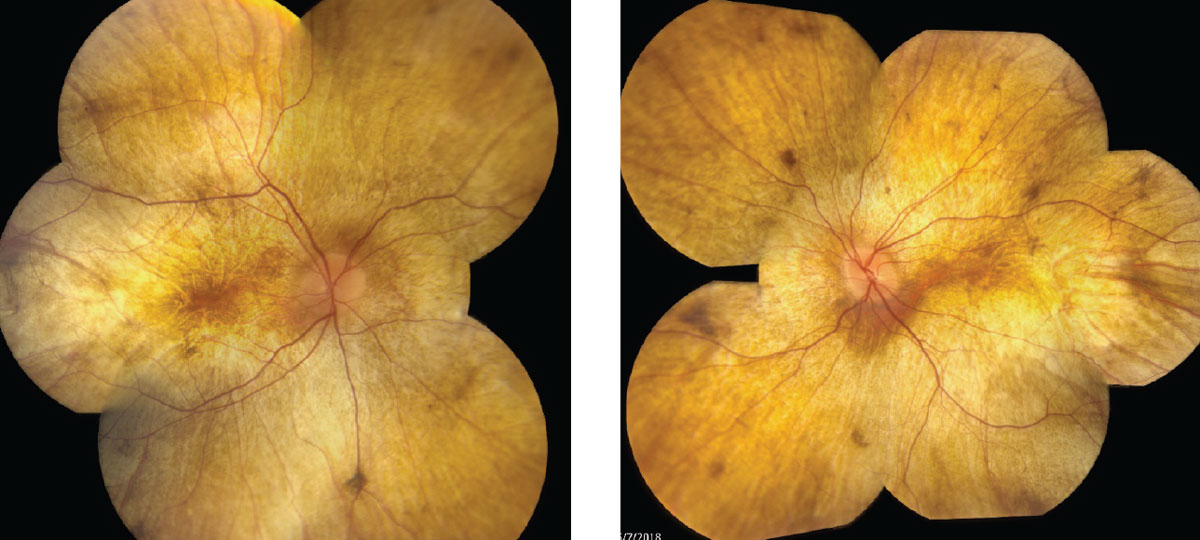 |
| Fig. 1. Fundus photos show our 34-year-old patient’s right, at left, and left eyes. Click images to enlarge. |
Take the Retina Quiz
1. Which best describes the fundus appearance in both eyes?
a. Diffuse retinal whitening, sparing the central macula.
b. Diffuse outer retinal atrophy with exposed sclera, sparing the macula.
c. Vascular attenuation, peripheral bony spicules and disc pallor.
d. Pisciform flecks with central retinal atrophy.
2. What is the correct diagnosis?
a. Gyrate atrophy.
b. Choroideremia.
c. Ocular albinism.
d. Pathologic myopia.
3. How would you describe the OCT findings?
a. Thin inner retina with mild cystoid macular edema.
b. Subretinal hyperreflectivity with a thin choroid.
c. Significant outer retinal atrophy with choroidal thinning and outer retinal tubulations.
d. Thin retina with abnormally thick choroid.
4. Based on the images and presentation, what is the appropriate next step?
a. Send for genetic testing.
b. Send for stroke evaluation.
c. Send for low vision evaluation.
d. Intravitreal anti-VEGF injection.
For answers, see below.
Diagnosis
Based on the clinical presentation and history, our patient was tentatively diagnosed with chorioderemia (CHM) and sent for genetic testing, which confirmed the diagnosis. CHM is an x-linked recessive chorioretinal dystrophy, which affects approximately one in 50,000 to 100,000 individuals.1,2 Since the development of genetic confirmation, more than 100 variations in the CHM gene have been discovered, opening the door for gene therapy.1,2
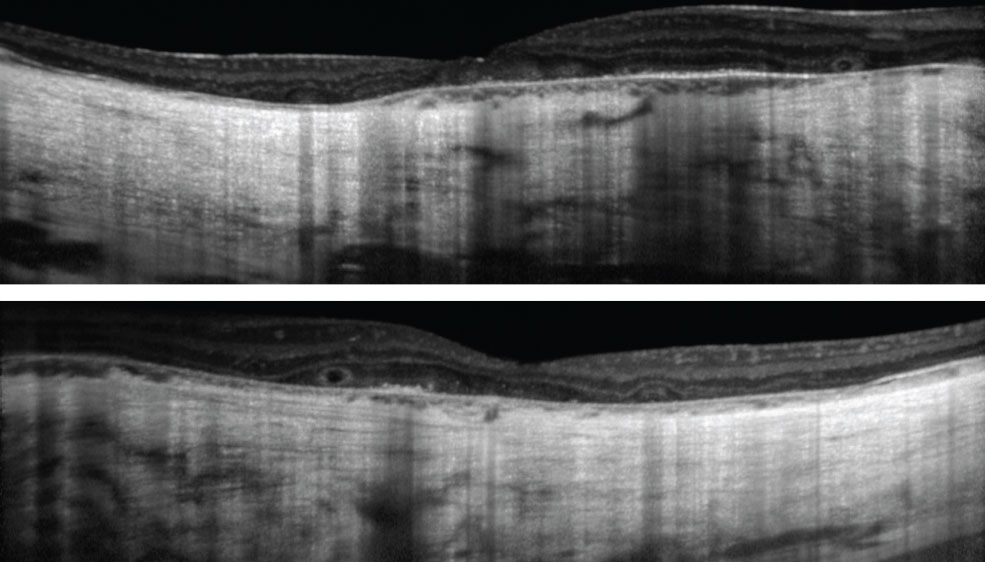 |
| Fig. 2. These OCT images show our patient’s right, at top, and left eyes. The patient has had visual complaints since childhood. Click images to enlarge. |
The CHM gene is responsible for the production of Rab escort protein-1 (REP-1), which is one of only two Rab escort proteins throughout the body. REP-1 helps to transport lipid membrane-bound structures (Rab) intracellularly, which plays a role in the transport of proteins from the Golgi apparatus to the outer segments in photoreceptors, and phagocytosis/degradation of outer segments shed by the RPE cells. Unfortunately, the other Rab escort protein cannot take over the function in the event of a defective REP-1.3 Ultimately, this results in degeneration to the photoreceptors (rods and cones), RPE and choriocapillaris.
Initial signs of diffuse RPE pigment clumping can be confused with bone-spicule pigment clumping associated with retinitis pigmentosa (RP), but can differentiated because RP pigment clumping is typically perivascular.4
Large areas of atrophy soon develop at the equator, extending centrally to the arcades, then the peripapillary region revealing sclera and choroidal vessels. The fundus appears stable, with an intact outer retina, RPE and choroid foveally until late-stage progression results in central atrophy as well.
Female carrier signs can vary from no changes to mild peripheral RPE changes/clumping, with or without atrophy. Other associated findings include posterior subcapsular cataracts and, rarely, choroidal neovascular membrane. Often, the pathophysiology and ocular signs directly correlate to visual symptoms.
In advanced CHM the fundus has a classic appearance of yellow/white sclera and large choroidal vessels with or without foveal sparing, depending on the degree of progression.
However, early in the disease, imaging can help in diagnosis and management (Table 1).
Table 1. Diagnostic Findings for CHM Staging. | ||
| Early | Advanced | |
| OCT | Slight central retinal and choroidal thickening Parafoveal outer retinal atrophy Cystoid macular edema | Central outer retinal atrophy Choroidal thickening Cystoid macular edema Outer retinal tubulations |
| ERG* | Reduced scotopic followed by reduced photopic | Extinguished |
| FAF** | Patchy hypo-AF with scalloped, distinct edge-sparing macula | Extensive hypo-AF involving macula and periphery |
| *Electroretinogram for carriers is normal, except in some symptomatic carriers, which may show reduced full-field ERG 30-Hz flicker response in 15% of symptomatic carriers.4 **Fundus autofluorescence is extremely useful in early diagnosis and predicting disease progression. Areas of hypo-autofluorescence may precede outer retinal atrophy, thus there may be minimal to no signs on fundus exam or photography. Carriers can show speckled Hyper-Autofluorescence resulting from mild RPE degeneration, thus lipofuscin accumulation.4 | ||
Prevalence
Due to the x-linked inheritance, patients are typically male. Females are rarely affected, but are often carriers. Initial symptoms involve nyctalopia (correlated to peripheral pigment clumping) beginning in the first decade of life, that progresses to peripheral vision loss (correlating to peripheral atrophy) throughout the second and third decades of life. Central vision is preserved until the fifth to seventh decades of life, when color vision, visual acuity or both, can rapidly deteriorate (correlating to foveal atrophy). A large variation in symptoms and disease progression exists, even between family members affected by the condition. Female carriers will be largely asymptomatic, or have mild changes to their night vision and peripheral vision correlating to peripheral clumping and atrophy.1
Follow Up
Genetic testing has become the standard for confirming a CHM diagnosis. As CHM has just a single gene mutation, clinicians hope to one day see development of a successful gene therapy treatment. Adeno-associated virus subtype 2 is used in ophthalmic gene therapy research, in large part due to its affinity for photoreceptors and RPE.
Our patient was subsequently enrolled in one of the current clinical trials involving gene therapy via subfoveal injection of an adeno-associated virus subtype 2 for chorioderemia. He is being followed closely to determine whether he experiences any improvement in his visual function.
| 1. Chan S, Bubela T, Dimopoulos I, et al. Choroideremia research: report and perspectives on the second international scientific symposium for choroideremia. Ophthalmic Genet. 2016 Sep;37(3):267-75. 2. Freund P, Sergeev Y, MacDonald I. Analysis of a large choroideremia dataset does not suggest a preference for inclusion of certain genotypes in future trials of gene therapy. Molecular Genetics & Genomic Medicine. 2016;4(3):344-58. 3. Imani S, Ijaz I, Shasaltaneh M, et al. Molecular genetics characterization and homology modeling of the CHM gene mutation: a study on its association with chorioderemia. Mutation Research. 2018;775(2):39-50. 4. MacDonald I, Hume S, Chan S, et al. Chorioderemia. GeneReviews. February 26, 2015. www.ncbi.nlm.nih.gov/books/NBK1337/. Accessed June 6, 2018. |
