 In our last column (“Hematology (Part 1): True Blood,” September 2011), we presented an overview of hematology—the study of the cellular elements of blood. Hematologic disorders result from excess or impaired production of blood cells, destruction of blood cells and abnormal function of existing blood cells.
In our last column (“Hematology (Part 1): True Blood,” September 2011), we presented an overview of hematology—the study of the cellular elements of blood. Hematologic disorders result from excess or impaired production of blood cells, destruction of blood cells and abnormal function of existing blood cells.
Common hematologic disorders include sickle cell hemoglobinopathy, anemias, bleeding and clotting disorders, and blood cancers. The anemias are a group of conditions in which red blood cells (RBCs) are insufficient in number or do not function properly.1,2
Hemoglobin (Hb) molecules in the RBCs transport and distribute oxygen throughout the body. Hemoglobinopathy results from structural differences in Hb produced by the body.
Hemoglobin is composed of heme (the portion containing iron) and globin (a protein made up of amino acid chains). Hb variants occur when mutations in the globin genes result in changes in the amino acids of the protein. Hundreds of variants have been identified, but just a small number are common and have clinical significance. Hb variants are inherited in an autosomal recessive manner; among the most important are HbS, HbC, HbE, HbSC and HbF.1-3
Sickle Cell Essentials
Sickle cell disease affects more than 72,000 Americans, primarily of African heritage. Persons of Arabian, Mediterranean, Caribbean, Indian, Asian, and South and Central American descent are also affected. Sickle cell disease occurs in about one in every 500 African-American births and one in every 1,000 to 1,400 Hispanic-American births. About 2 million Americans, or one in 12 African-Americans, carry the sickle cell trait.2,3
Sickle cell disease is an inherited disorder that affects RBCs. Normal red cells are soft, smooth and round, enabling them to move easily through blood vessels. Sickle cell anemia is a chronic hemolytic condition in which the Hb protein is abnormal, causing the RBCs to be rigid, sticky, sickle-shaped and fragile. These altered RBCs obstruct the circulation because they are unable to flow through small blood vessels. The most common types of sickle cell disease are SS, SC and S beta thalassemia.3,4
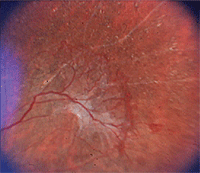
1. Proliferative sickle retinopathy: “sea fan” neovascularization in a patient with sickle cell anemia.
Occlusion of blood vessels prevents the delivery of oxygen to tissues and organs. Sickle cells die after just 10 to 20 days. Because they cannot be replaced fast enough, the blood is chronically short of RBCs, leading to anemia.3-5 Patients with sickle cell anemia experience a wide range of symptoms, including periodic episodes of pain.
The Clinical Profile
The clinical course of sickle cell anemia does not follow a single pattern. Whereas some patients experience only mild symptoms, others live with debilitating complications. Fatigue, paleness and shortness of breath all may occur. Most acute complications of sickle cell disease are related to blood vessel occlusion.
Painful crises, caused by ischemic pain in organs, can occur anywhere in the body. The pain is found most commonly in the extremities, chest, abdomen and back. A patient may experience up to as many as 15 crises per year. The pain may last from a few hours to several weeks.3-5
Vaso-occlusion in the pulmonary circulation can result in acute chest syndrome, which involves fever, cough, sputum production, dyspnea or hypoxia.4,5 Neurologic events are a major cause of morbidity in patients with sickle cell disease. Acute, large vessel brain occlusions can occur in children; however, such strokes rarely occur in adults.4,5
The spleen is also a site in which recurrent sickling uniformly occurs. Patients become functionally asplenic from repeated infarctions of the microvasculature. This increases their susceptibility to infections with encapsulated organisms. Acute infection remains a significant cause of death.3,5
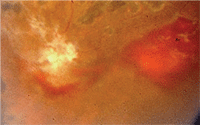
2. Non-proliferative sickle retinopathy: salmon patch hemorrhage in another patient.
Renal disease and pulmonary failure are two leading causes of death in adult patients. Other long-term complications include retinopathy (figures 1 and 2), chronic skin ulcers, avascular necrosis and liver dysfunction.
You should suspect sickle cell disease in patients of color with hemolytic anemia and repeated episodes of joint, bone or thoracic pain.
General screening is performed with a routine complete blood count with differential and a Sickledex test. More specific testing might include hemoglobin electrophoresis or DNA analysis.3-5
Public Health Measures
Every U.S. state now mandates testing for sickle cell anemia as part of its newborn-screening programs. This test is performed at the same time and from the same blood sample as other routine newborn screening tests. Hb electrophoresis is the most widely used diagnostic test. If it shows the presence of sickle hemoglobin, a second blood test is performed to confirm the diagnosis. The second test should be done as soon as possible and within the first few months of life.4,6,7
It is now possible to diagnose sickle cell disease before birth. This is done using a sample of amniotic fluid or placental tissue. Testing before birth can be done as early as 10 weeks into the pregnancy. This testing looks for the sickle hemoglobin gene, rather than the abnormal hemoglobin that the gene triggers.6,7
Current and Emerging Treatments
Although there is no cure for sickle cell anemia, providers can do a great deal with supportive therapies to help their patients. Patients need ongoing treatment, even when they are not having a painful crisis. Supplementation with folic acid is essential for producing red blood cells because they are turned over rather quickly.1,3,7
Manifestations of non-proliferative or background sickle retinopathy include: • Venous tortuosity.
Proliferative sickle retinopathy, the most severe ocular change in
sickle cell disease, is progressive. Goldberg classified it using five
stages: • Stage 1: Peripheral arteriolar occlusions.
Patients with sickle cell anemia need to keep all of their immunizations up to date. Antibiotics and vaccines are given to prevent bacterial infections, which are common in children. Painful crises are treated with fluid intake, oxygen supplementation and analgesics. In addition, increasing the number of normal RBCs through transfusions may be beneficial. Transfusions can also be used to treat spleen enlargement in children.3,7
Non-proliferative vs. Proliferative Sickle Retinopathy8
• Salmon-patch hemorrhages.
• Iridescent spots and glistening refractive bodies in the schisis cavity.
• The black sunburst.
• Stage 2: Arteriolar-venular anastomosis.
• Stage 3: Neovascular proliferation.
• Stage 4: Vitreous hemorrhage.
• Stage 5: Retinal detachment.
Regular transfusion therapy can help prevent recurring strokes in children at high risk. Patients who present with infection should receive antibiotic treatment. Hydrea (hydroxyurea, Bristol-Myers Squibb), an agent that increases the concentration of hemoglobin F, reduces the incidence of vaso-occlusive crises.1,7
Bone marrow or stem cell transplants may eventually be used to help people with sickle cell anemia. However, transplantation carries many risks, including infection, rejection and graft-versus-host disease.1,5,7 So, it is not a likely option for most patients. In addition, patients are often unable to find well-matched donors.
Hemoglobinopathies, including sickle cell anemia, are a major public health challenge in the United States. Team-based health care is critical, including regular visits to the optometrist. Proper nutrition and hygiene, appropriate rest, protection against infection and avoidance of stress all are important in maintaining wellness and preventing complications.
1. Rose M, Berliner N. Disorders of red blood. In: Andreoli TE, Carpenter CCJ, Griggs RC, Benjamin I. Cecil’s Essentials of Medicine. 7th ed. Philadelphia: Saunders Elsevier; 2007:497-508.
2. Steinberg MH. Sickle cell trait. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL (eds). Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge, UK: Cambridge University Press; 2001:811-30.
3. American Society of Hematology. Blood: The Vital Connection. Available at:
www.hematology.org/Patients (accessed October 2, 2011).
4. Pagana KD, Pagana TJ. Mosby’s Diagnostic and Laboratory Test Reference. 8th ed. St. Louis: Mosby, Inc.; 2007:290-95.
5. Stiene-Martin EA, Lotspeich-Steininger CA, Koepke JA. Clinical Hematology: Principles, Procedures, Correlations. 2nd ed. Philadelphia: Lippincott-Raven Publishers; 1998.
6. Sickle Cell Disease Guideline Panel. Sickle cell disease: screening, diagnosis, management, and counseling in newborns and infants. Clinical Practice Guideline no. 6, Rockville, MD: Agency for Health Care Policy and Research, U.S. Public Health Service, 1993, AHCPR Publ. No. 93-0562.
7. Schnog JB, Duits AJ, Muskiet FA, et al. Sickle cell disease; a general overview. Neth J Med 2004 Nov;62(10):364-74.
8. Hampton R. Ophthalmologic manifestations of sickle cell disease. Available at:
http://emedicine.medscape.com/article/1918423-overview#a30. Accessed November 1, 2011.
