He reported no family history of ocular disease or visual problems. His systemic history was significant for type 2 diabetes mellitus, as well as uncontrolled hypertension that had resulted in kidney failure. He was also diagnosed with anemia approximately 11 months earlier.
On examination, his best-corrected visual acuity measured 20/20 OD and 20/30 OS. Confrontation fields showed generalized constriction 360° OU. His pupils were equally round and reactive, with no evidence of afferent defect OU.
The anterior segment was remarkable for posterior subcap-sular cataracts (PSC) OU, with trace amounts of nuclear and cortical changes. His IOP measured 15mm Hg OU. His blood pressure was 176mm Hg/112mm Hg.
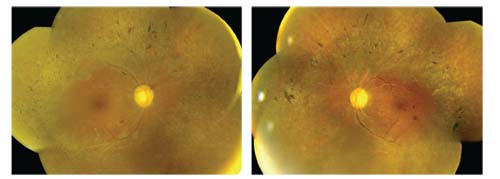
Dilated fundus examination of both eyes revealed symmetric posterior pole and mid-peripheral changes (figures 1 and 2). We also obtained an SD-OCT scan of both eyes, which showed very mild cystoid macular edema (CME).
 |
|
1,2. Dilated fundus images of our patient (OD left, OS right) revealed several significant findings. What do you notice?
|
Take the Retina Quiz
1. Based upon the findings observed along the arcades, which underlying condition does our patient likely have?
a. Syphilis.
b. Retinitis pigmentosa (RP).
c. Intermediate uveitis.
d. Sarcoidosis.
2. What do the posterior pole changes likely represent?
a. Diabetic retinopathy.
b. Hypertensive retinopathy.
c. Retinal artery occlusions.
d. All the above.
3. Which ancillary test would be of LEAST diagnostic value for this patient?
a. MRI/CT of the brain and orbits.
b. Electroretinogram (ERG).
c. Fluorescein angiography.
d. Genetic testing.
4. What is the most appropriate treatment for his CME?
a. Acular (ketorolac trometamine, Allergan).
b. Anti-VEGF injection.
c. Acetazolamide.
d. None of the above.
5. How should this patient be managed?
a. Immediate referral for blood pressure control.
b. Visual fields and ERG testing.
c. Blood work-up, including ACE and FTA-ABS.
d. Topical prostaglandin therapy.
(Answers at end of article).
Discussion
Because our patient presented with a multitude of clinical findings, it was especially difficult to establish a definitive diagnosis. The numerous sclerotic vessels, flame hemorrhages and bilateral disc hemorrhages are indicative of severe hypertensive retinopathy. Indeed, when we took his blood pressure, it was extremely elevated––likely a hypertensive emergency.
Additionally, he exhibited microaneurysms and blot hemorrhages (OS > OD) that were consistent with diabetes. Further, the pigment spiculing noted along the arcades and the “moth-eaten” retinal degenerative changes were indicative of RP. Other characteristic changes associated with RP included myopia, arteriole attenuation, waxy pallor (possibly drusen), PSC, CME and vitreous cells. It is important to note that PSC occur in nearly 50% of patients who are in the later stages of RP.1
Retinitis pigmentosa encompasses a clinically and genetically varied group of hereditary retinal disorders, and is one of the most common causes of retinal degeneration. Its overall prevalence is approximately one in 4,000 individuals. There is no racial predilection, but RP is more common in males. Although the term “retinitis” is indicative of underlying inflammation, histopathology shows no evidence of an inflammatory response. Instead, affected individuals exhibit photoreceptor apoptosis. Most RP cases result in the death of rod photoreceptors, which severely impairs vision in dim lighting.
Some forms of RP may affect the cones before the rods (termed cone-rod dystrophy). The cone-rod variant commonly presents with late-onset night blindness, less pigment deposition, better preservation of visual fields and more recordable ERG patterns than the rod-cone variant (although there is tremendous variability in the presentation). Most RP patients with the cone-rod variant don’t complain of night blindness until the visual field is constricted to 10°.
The various forms of RP all have a genetic component, so most patients will report some type of family history. This is not always the case, however. Infrequently, patients with no family history of RP may present with a sporadic mutation. This can confound the diagnosis, making it difficult to differentiate the condition from inflammatory or infectious diseases. These sporadic retinitis pigmentosa cases, also referred to as “isolated” or “simplex” RP, occur in up to 50% of all affected patients.1
Management of RP should include vitamin A supplementation, as well as avoidance of excess vitamin E intake. In a randomized, controlled study, adult RP patients showed a significant reduction in disease progression following daily supplementation with 15,000IU of vitamin A over a four- to six-year period.2 While the researchers determined that vitamin A protects the remaining cones, they suggested that vitamin E inhibits the efficacy and distribution of vitamin A uptake.
Before initiating supplementation, patients should undergo fasting serum vitamin A and liver function profiles. Keep in mind that pregnant women should not take 15,000IU of vitamin A per day because of an increased risk of birth defects. Further, those with a history renal disease or kidney transplant should not take this large of a dosage because of an elevated risk of toxicity secondary to increased absorption. Finally, it is important to note that patients who take doxycycline may be at an increased risk for intracranial pressure following excess vitamin A supplementation.
RP patients with concomitant CME can be treated with topical NSAIDs, steroids and carbonic anhydrase inhibitors (CAIs). If topical medications fail, oral CAIs (e.g., 250mg acetazolamide BID for six weeks) or periocular/intravitreal steroids can be used.
Currently, gene therapy for RP using recombinant adeno-associated viral vectors has been both safe and effective in animal models.3 However, based upon preliminary results, researchers have high expectations for its future application in humans.
We educated our patient about his new diagnosis of RP, as well as advised him and his children to undergo further genetic testing. Such a measure would help the family prepare for potential ocular changes seen in the future.
More importantly, however, our patient’s blood pressure required urgent control. Given this and his retinal vascular changes, he clearly was in an emergency situation. We instructed him to see his primary care physician that day, and scheduled him for visual fields and ERG testing within the next month.
Thanks to Stephanie Frankel, OD, optometric resident at Bascom Palmer Eye Institute in Miami, for contributing this case.
Quiz Answers: 1) B; 2) D; 3) A; 4) D; 5) A.
1. Alexander L. Primary Care of the Posterior Segment, 2nd ed. New York: Appleton and Lange; 1994.
2. Berson EL. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. Arch Ophthalmol. 1993 Jun;111(6):761-72.
3. Fahad I, Al-Saikhan. The gene therapy revolution in ophthalmology. Saudi J Ophthalmol. 2013 April;27(2):107-11.
