Prior to the 1990s, few geneticists were interested in identifying glaucoma genes. After all, the disease had a late age of onset, and they estimated that the heritability of glaucoma was as low as 13%.1
In reality, however, glaucoma is the second most prevalent cause of bilateral blindness in the world, behind diabetes.2 It affects more than 2.2 million people in the United States alonea number projected to increase by 50% by the year 2020.3 It is a complex, heterogeneous group of diseases that cause ganglion cell death and optic neuropathy.
Since the 1990s, discovery of glaucoma genes and the proteins they produce has improved our understanding of the pathophysiology of this complex group of diseases. (See Predicting Glaucoma Based on Proteomics, below.) Once the first locus was discovered in 1993, it sparked an increased interest in genetics. This interest was also sparked by the increased risk (9.2) of patients developing glaucoma themselves.4 Researchers also realized that this knowledge would lead to the development of genetic tests and perhaps new approaches to treatment.
|
Predicting Glaucoma Based on Proteomics |
| Proteomics is the study of the proteins that genes produce and how these proteins function. Proteomic profiling studies in glaucoma are currently helping to identify which proteins in the blood, when over- or under-expressed, are associated with the development of various types of glaucoma. Some of these proteins have been used as a marker for inflammatory disease but also have been found to be associated with glaucoma. One such protein, SAA (serum amyloid A), causes amyloid deposition in tissues. Researchers found that an over-expression of SAA in the trabecular meshwork of glaucoma patients leads to decreased aqueous outflow in humans and increased IOP in mice.42 The future in early glaucoma diagnosis may one day lie in a simple blood test. This test may not look for glaucoma-associated genetic mutations but will test for the presence of proteins which, when over-or under-expressed, are associated with the development of glaucoma. These proteins may appear in the blood years before any structural or functional signs of glaucoma. Proteomics also opens up the possibility for future prevention of glaucoma by devel-oping drugs to reduce the over-expression or increase the under-expression of these proteins. |
Here, we will discuss what is currently known about human glaucoma genes.
Primary Open-Angle Glaucoma
The forms of primary open-angle glaucoma (POAG) that may be associated with a genetic mutation are adult-onset POAG, juvenile-onset POAG (glaucoma before age 40) and low- or normal-tension POAG.
Researchers discovered the first primary open-angle glaucoma locus in 1993.10 The mapping of this first glaucoma locus incited the identification of many other glaucoma gene loci within the next six years. So far, researchers have mapped at least 15 genetic loci for POAG but have actually identified or characterized only a small number of genes.
The first POAG locus to be identified was in pedigrees with juvenile- and adult-onset glaucoma. It was labeled GLC1A and found in the long arm of chromosome 1. (See What is a Locus"?" below.) Eventually, the myocilin, or MYOC, gene was identified at this locus in 1997. 11
|
What Is a Locus? |
| The first step to characterizing a gene involves the discovery of specific loci, or sites, on chromosomes that are common to individuals (in a large pedigree) who are affected by a specific disease. These loci are not found in family members who do not have the disease. A locus for glaucoma consists of (in this order): The symbol GLC, which is used to identify primary glaucoma loci. For example, GLC1A represents the locus for primary open-angle (1) glaucoma (GLC). The A indicates that this was the first POAG locus that researchers identified. |
The second POAG locus to be identified, GLC1B, is mapped to chromosome 2. 15 Most patients who have mutations in this locus have normal-tension glaucoma that develops after age 40.
GLC1C, the third POAG locus to be discovered, is mapped to chromosome 3. It is associated with POAG that is characterized by high intraocular pressure (IOP), late age of onset (ages 38 to 80) and a moderate response to IOP-lowering medications.16,17
GLC1D, the fourth locus to be discovered in the late 1990s, is also associated with a form of adult-onset POAG associated with high IOP. It has been mapped to the long arm of chromosome 8.18
Researchers have not yet identified the genes or proteins produced by mutations in the GLC1B, GLC1C and GLC1D loci.
|
|
| Here is pigment in the angle in pigment dispersion glaucoma. Researchers mapped the first human pigment dispersion glaucoma locus to chromosome 7 but have not yet identified a gene. So, we cannot currently predict which patients who have pigment dispersion syndrome will develop pigment dispersion glaucoma. |
Other reports suggest that, depending on ethnicity factors, OPTN may also play a role in high-tension glaucoma and juvenile open-angle glaucoma.20-24 Research has not clearly defined the role OPTN plays in low-tension glaucoma, although it may help protect the ganglion cells from factors that induce apoptosis, or programmed cell death.25

GLC1F, the sixth discovered locus for POAG, maps to the short arm of chromosome 7. It is associated with elevated IOP (22mm Hg to 38mm Hg).26 No gene has yet been identified.
GLC1G, the seventh locus to be discovered, is located on chromosome 5 and on the third adult-onset POAG gene, called WDR36, to be identified.27 It is associated with high- and low-tension forms of glaucoma. Like OPTN, this gene codes for a protein that is present in both ocular and non-ocular tissues. And, like OPTN, this genes role in the pathogenesis of glaucoma is not yet clearly defined.
Most recently, two other primary open-angle glaucoma loci have been identified: GLC1H, on chromosome 2associated with adult-onset glaucomaand GLC1Iassociated with earlier adult onset glaucoma.28
Besides the open-angle glaucoma genes identified thus far, there are susceptibility genes that may contribute to POAG. These genes affect the clinical course of POAG within an individual. Thus, family members who have the same mutation may have different severity of disease.
Studies are underway to identify the location of these susceptibility genes using sibling pairs among many families. The development of POAG can also vary within families who have the same glaucoma mutations. So-called modifier genes may affect the expression of some glaucoma mutations. The role of modifier genes currently is not well understood.
Pigment Dispersion Syndrome
Pigment dispersion syndrome, usually found in young, myopic Caucasians, is characterized by the release of pigment from the iris. This pigment can coat the lens, lens zonules, trabecular meshwork and corneal endothelium.29 It is usually suspected when an examination turns up endothelial pigment or Krukenbergs spindle.
Transillumination of the pupil before dilation, possibly due to loss of pigment from the iris, is a sign of this syndrome. Gonioscopy reveals dense pigment in the trabecular meshwork.
Gonioscopy may also reveal a concave iris, but ultrasound biomicroscopy (UBM) or another anterior segment imaging device is necessary to confirm this. A concave-shaped iris may result in the iris rubbing against the lens, which in turn releases pigment. Laser iridectomy can help flatten the iris.
Pigmentary dispersion is more prevalent in myopes because these patients have a concavity to their peripheral iris. Pigment dispersion is also seen more in younger patients, as age-related anatomical changes in the eye cause the iris to lift off the lens zonules. (So, as these patients age, less pigment is dispersed because of these anatomical changes.)
|
Fast Facts |
|
Transillumination of the pupil before dilation, possibly due to loss of pigment from the iris, is a sign of pigment dispersion syndrome. |
Pseudoexfoliation Glaucoma
Pseudoexfoliation, which is the production and accumulation of a fibrillar extracellular whitish material, can occur in ocular and nonocular tissues. The ocular tissues are comprised of the trabecular meshwork, the iris, and most notably, the lens and lens zonules.
This white, flaky material can best be seen on the lens surface through a dilated pupil but in some cases, may also be observable along the pupillary margin of an undilated pupil. Because this material does not represent true exfoliation of the lens capsulenow called capsular delamination of the lens (XS) the process is referred to as pseudoexfoliation.
Pseudoexfoliative syndrome (XFS or PEX) affects several sites in the eye or elsewhere in the body. It leads to pseudoexfoliation glauco- ma in 40% to 50% of patients. 31
Although this condition has a high prevalence in individuals of Scandinavian descent, researchers have not yet mapped any genetic loci, nor have they identified any genes associated with this form of glaucoma.
However, in studies of twins, inheritance in two-generation families increased risk in individuals who had affected relatives, and prevalence differences in specific populations are suggestive of autosomal dominant, X-linked and mitochondrial inheritance. 32-39
|
|
| Pseudoexfoliation is the production and accumulation of a fibrillar extracellular whitish material in the ocular tissues, which |

Congenital Glaucoma
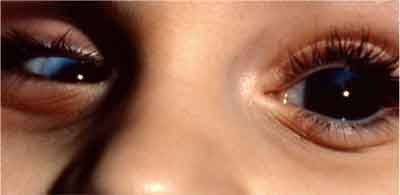
This form of glaucoma differs from juvenile-onset glaucoma in that it is an autosomal-recessive inherited, severe form of glaucoma that occurs before age 1. Most cases are bilateral.
Presenting clinical symptoms include epiphora, obvious photophobia and blepharospasm. Examination of the cornea may reveal corneal cloudiness and breaks in Descemets membrane, called Haabs striae, due to corneal stretching from the increased IOP. Also, the corneal diameter increases (megalocornea) so that the eye or eyes may look like marbles.
Gonioscopy reveals malformation of the trabecular meshwork. Mutations in the gene that codes for the CYP1B1 (cytochrome P450 1B1) protein (locus GLC3A on chromosome 2) have been detected in numerous families from different countries where individuals have congenital glaucoma.40
Also, mutations in this gene are responsible for abnormal ocular development. However, most cases are sporadic, with no family history.
Developmental Glaucoma
The developmental glaucomas are diseases in which glaucoma is part of a syndrome. These syndromes include Axenfelds anomaly (anteriorly displaced Schwalbes line with peripheral iris strands), Riegers anomaly (same as Axenfelds but with iris atrophy), aniridia, autosomal-dominant irideogoniodygenesis anomaly and Peters anomaly (corneal opacification, iris adhesions and cataract).
These congenital anomalies of the anterior segment can subsequently result in the development of glaucoma. There may be malformations evident in other parts of the body as well, such as in the teeth, facial bones and heart.
Mutations in at least four different genes can lead to all these anomalies. These four genes are PITX2, PAX6, FOXC1 and CYP1B1. Mutations in these genes do not necessarily result in the same phenotype. Each is affected by environmental factors as well. For example, mutations in the PITX2 gene can give rise to Axenfelds anomaly, Riegers syndrome or Peters anomaly, all within the same family.41 (See Summary of the Loci and Genes Associated With Various Forms of Glaucoma, above.)
|
How to Order Genetic Testing |
|
If your patient has a strong family history of early-onset (juvenile) open-angle glaucoma, you may want to offer genetic testing for mutations or variants in the MYOC gene. |
Genetic Testing
As more genes and mutations are identified, should all family members be screened in glaucoma pedigrees to predict the clinical course or severity of the disease? Currently, the answer is no. The relationship between the genetic mutation and resulting disease severity is not yet clear, especially in POAG. So, testing for glaucoma mutations is currently not clinically useful for most forms of glaucoma.
However, you may want to consider offering testing of the MYOC gene to families who have a history of severe early-onset (juvenile) open-angle glaucoma (see How to Order Glaucoma Genetic Testing, above).
As for mass screenings of large populations, we must wait for the discovery of more glaucoma-causing genes and the development of highly sensitive and specific, cost-effective tests that screen for all the mutations in these genes.
The identified genes mentioned here account for only a small number of the overall glaucoma cases we see clinically. In reality, most glaucoma we see is caused by multiple factors. In fact, the interaction of multiple genetic sites likely contributes to disease development.
In the future, the discovery of genes and the proteins they produce will help us differentially diagnose the various forms of glaucoma earlier, predict disease severity, and develop prophylactic treatment.
|
|
| Presenting clinical symptoms of congenital glaucoma include megalocornea in this patients left eye, which is the diameter increase of the cornea. Microcornea is seen in the right eye. |
Dr. Bass is a distinguished teaching professor at the State University of New York State College of Optometry. She is an instructor in the posterior segment disease course in the professional program, and she teaches in the glaucoma, retina and cornea clinic.
Next month: In the final article in this series, Dr. Bass discusses genetic disease of the cornea.
1. Teikari JM. Genetics factors in open-angle (simple and capsular) glaucoma. A population-based twin study. Acta Opthalmologica (Copenh) 1987 Dec;65 (6):715-20.
2. Diabetes Programme. Welcome to the Diabetes Programme. World Health Organization. www.who.int/
diabetes/en/ (Accessed 2 Aug, 2006).
3. Friedman DS, Wolfs RC, OColmain BJ, et al. Prevalence of open-angle glaucoma among adults in the United States. Arch Ophthalmol 2004 Apr;122(4):532-8.
4. Wolfs RC, Klaver CC, Ramrattan RS, et al. Genetic risk of primary open-angle glaucoma. Population-based familial aggregation study. Arch Ophthalmol 1998 Dec;116(12): 1640-5.
5. Wilson MR. Epidemiological features of glaucoma. Int Ophthalmol Clin 1990 Summer;30(3):153-60.
6. Weale R. Is the season of birth a risk factor in glaucoma? Brit J Ophthalmol 1993 Apr;77(4):214-7.
7. Klein BE, Klein R, Lee KE. Heritability of risk factors for primary open-angle glaucoma: the Beaver Dam Eye Study. Invest Ophthalmol Vis Sci 2004 Jan;45(1):59-62.
8. Chang TC, Congdon NG, Wojciechowski R, et al. Determinants and heritability of intraocular pressure and cup-to-disc ratio in a defined older population. Ophthalmology 2005 Jul;112(7):1186-91.
9. Toh T, Liew SH, MacKinnon JR, et al. Central corneal thickness is highly heritable: the twin eye studies. Invest Ophthalmol Vis Sci 2005 Oct;46(10):3718-22.
10. Sheffield VC, Stone EM, Alward WL, et al. Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nat Genet 1993 May;4(1):47-50.
11. Stone EM, Fingert JH, Alward WL, et al. Identification of a gene that causes primary open angle glaucoma. Science 1997 Jan 31;275(5300):668-70.
12. Polansky JR, Fauss DJ, Chen P, et al. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica 1997;211(3):126-39.
13. Wiggs JL, Allingham RR, Vollrath D, et al. Prevalence of mutations in TIGR/myocilin in patients with adult and juvenile primary open-angle glaucoma. Am J Hum Genet 1998 Nov;63(5):1549-52.
14. Shimizu S, Lichter PR, Johnson AT, et al. Age-dependent prevalence of mutations at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol 2000 Aug;130(2):165-77.
15. Stoilova D, Child A, Trifan OC, et al. Localization of a locus (GLC1B) for adult-onset primary open-angle glaucoma to the 2cen-q13 region. Genomics 1996 Aug15;36(1):
142-50.
16. Wirtz MK, Samples JR, Kramer PL, et al. Mapping a gene for adult-onset primary open-angle glaucoma to chromosome 3q. Am J Hum Genet 1997 Feb;60(2):296-304.
17. Kitsos G, Eiberg H, Economou-Petersen E, et al. Genetic linkage of autosomal dominant primary open angle glaucoma to chromosome 3q in a Greek pedigree. Eur J Hum Genet 2001 Jun;9(6):452-7.
18. Trifan OC, Traboulsi EI, Stoilova D, et al. A third locus (GLC1D) for adult-onset primary open-angle glaucoma maps to the 8q23 region. Am J Ophthalmol 1998 Jul;126(1):17-28.
19. Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002 Feb 8;295(5557):1077-9.
20. Funayama T, Ishikawa K, Ohtake Y, et al. Variants in opti-neurin gene and their association with tumor necrosis factor-alpha polymorphisms in Japanese patients with glaucoma. Invest Ophthalmol Vis Sci 2004 Dec;45(12):4359-67.
21. Umeda T, Matsuo T, Nagayama M, et al. Clinical relevance of optineurin sequence alterations in Japanese glaucoma patients. Ophthalmic Genet 2004 Jun;25(2):91-9.
22. Willoughby CE, Chan LL, Herd S, et al. Defining the pathogenicity of optineurin in juvenile open-angle glaucoma. Invest Ophthalmol Vis Sci 2004 Sep;45(9):3122-30.
23. Fuse N, Takahashi K, Akiyama H, et al. Molecular genetic analysis of optineurin gene for primary open-angle and
normal tension glaucoma in the Japanese population. J Glaucoma 2004 Aug;13(4):299-303.
24. Fan BJ, Wang DY, Fan DS, et al. SNPs and interaction analyses of myocilin, optineurin and apolipoprotein E in primary open angle glaucoma patients. Mol Vis 2005 Aug 29; 11:625-31.
25. DeMarco N, Buono M, Troise F, Diez-Roux G. Opti-neurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem 2006 Jun 9;281(23):16147-56. Epub 2006
Mar 28.
26. Wirtz MK, Samples JR, Rust K, et al. GLC1F, a new primary open-angle glaucoma locus, maps to 7q35-q36. Arch Ophthalmol 1999 Feb;117(2):237-41.
27. Monemi S, Spaeth G, DaSilva A, et al. A1. Identification of a novel adult-onset primary open-angel glaucoma (POAG) gene on 5q22.1. Hum Mol Genet 2005 March 15;14(6):725-33 Epub 2005 Jan 27.
28. Allingham RR, Wiggs JL, Hauser ER, et al. Early
adult-onset POAG linked to 15q11-13 using ordered subset analysis. Invest Ophthalmol Vis Sci 2005 Jun 46(6):2002-5.
29. Sowka J. Pigmentary dispersion syndrome and pigmentary glaucoma. Optometry 2004 Feb;75(2):115-22.
30. Andersen JS, Pralea AM, DelBono EA, et al. A gene re-sponsible for the pigment dispersion syndrome maps to chromosome 7q35-q36. Arch Ophthalmol 1997 Mar;115(3): 384-8.
31. Karger RA, Jeng SM, Johnson DH et al. Estimated incidence of pseudoexfoliative syndrome and pseudoexfoliation glaucoma in Olmsted County, Minnesota. J Glaucoma 2003 Jun;12(3):193-7.
32. Damji KF, Bains HS, Stefansson E, et al. Is pseudoexfoliation syndrome inherited? A review of genetic and nonge-netic factors and a new observation. Ophthalmic Genet 1998 Dec;19(4):175-85.
33. Gottfredsdottir MS, Sverrison T, Musch DC, Stefansson E. Chronic open-angle glaucoma and associated ophthalmic findings in monozygotic twins and their spouses in Iceland. J Glaucoma 1999 Apr;8(2):134-9
34. Hardie JG, Mercieca F, Fenech T, Cuschieri A. Familial pseudoexfoliation in Gozo. Eye 2005 Dec;19(12):1280-5.
35. Forsius H, Forsman E, Fellman J, Eriksson AW. Exfoliation syndrome: frequency, gender distribution and association with climatically induced alterations of the cornea and conjunctiva. Acta Ophthalmol Scand 2002 Oct;80(5):478-84.
36. Orr AC, Robitaille JM, Price PA, et al. Exfoliation syndrome: clinical and genetic features. Ophthalmic Genet 2001Sept;22(3):171-85.
37. Alligham RR, Loftsdottir M, Gottfredsdottir, et al. Pseudoexfoliation syndrome in Icelandic families. Br J Ophthalmol 2001 Jun;85(6):702-7.
38. Damji KF, Bains HS, Amjadi K, et al. Familial occurrence of pseudoexfoliation in Canada. Can J Ophthalmol 1999 Aug;34(5):257-65.
39. Kozobolis VP, Detorakis ET, Sourvinos G, et al. Loss of heterozygosity in pseudoexfoliation syndrome. Invest Ophthalmol Vis Sci 1999 May;40(6):1255-60.
40. Stoilov, I Akarsu AN, Sarfarzi, M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet 1997 Apr;6(4):641-7.
41. Honkanen RA, Nishimura DY, Swiderski RE, et al. A family with Axenfeld-Reiger syndrome and Peters anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol 2003 Mar;135:368-75.
42. Wang WH, McNatt LG, Pang IH, et al. Increased expression of serum amyloid A in human glaucoma and effect on IOP. IOVS Suppl 2006:1545.
