As primary eye care practitioners working with a rapidly aging population, our role in glaucoma management is expanding. Unfortunately for patients and practitioners, the pathogenesis of glaucoma is not yet fully understood. A multitude of causal mechanisms have been proposed, including: low cerebrospinal fluid pressure leading to laminar cribrosa non-support, impaired microcirculation and vascular dysregulation, altered immunity, excitotoxicity, oxidative stress and secondary neurodegeneration of retinal neurons and cells from primary neural pathological processes.1,2
As we close in on its origin, we may get closer to discovering how to defeat the disease. But for now, what we do know is that intraocular pressure (IOP) is the sole risk factor we can currently modify to alter the characteristic death of retinal ganglion cells seen in glaucoma patients.2-6 Often, IOP modification requires surgical intervention.
Extensive scientific research into a non-surgical option has led scientists to pharmacologically target the cells of the trabecular meshwork (TM) to facilitate aqueous outflow.7 The pharmacological mechanism involved relates to inhibition of Rho GTPase proteins, most notably RhoA, which exists in significantly elevated levels in glaucomatous optic nerve heads (ONH), suggesting they play a role in the disease’s pathophysiology.8
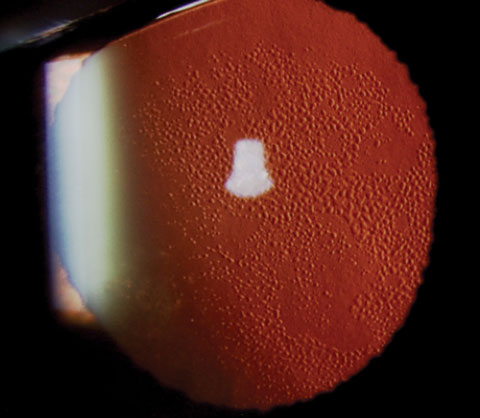 |
| A case of Fuch’s dystrophy shown in retroillumination. Research now demonstrates the ability of ROCK inhibitor eye drops, in combination with transcorneal freezing, to decrease central corneal thickness and recover corneal clarity in Fuch’s cases. |
Clinical research involving Rho-kinase inhibitors (known as ROCK inhibitors) shows promise, one day, to provide additional agents to the topical glaucoma-management armamentarium available in the United States.
TM Outflow Pathway
To understand how ROCK inhibitors work, you first need a little background on the operations of the TM and how, on a cellular level, it provides the mechanism for aqueous outflow.
The TM outflow pathway accounts for up to 90% of aqueous outflow under normal physiological conditions.9-11 Anatomically, the TM can be separated into distinct regions based on location and function. The regions of the uveal and corneoscleral meshwork consist of arrays of lamellae, which are, themselves, comprised of fenestrated collagen beams. These beams are covered by endothelial-like cells, with loose extracellular matrix (ECM) that occupies the spaces between the cells of the adjacent beams.12 These spaces decrease in size and the lamellae become flatter as they transition into the area known as the juxtacanalicular (JCT), or cribriform, region. This JCT is composed of cells embedded in a dense network of ECM with narrow intercellular spaces.13 The ECM provides a channel for aqueous humor (AH) to cross the JCT and exit the anterior chamber through Schlemm’s canal, where the AH is eventually drained into the venous circulation. The ECM is an active structure, possessing many bioactive molecules that influence outflow resistance. This activity in the extracellular environment is linked to alterations in the intracellular actin cytoskeleton and vice versa.13 Resistance to aqueous flow in normals is greatest in the JCT region and/or the inner wall of Schlemm’s canal.9
Regulation of aqueous outflow in this pathway is primarily controlled by the interaction of two cell types in the JCT region: cells of the TM and Schlemm’s canal endothelia. Cells of the TM express smooth muscle-like properties including contractility, electro-mechanical characteristics and expression of actin and myosin specific to smooth muscle tissue.14 This highly structured cellular actomyosin system affects the overall contractile tone of the tissue, influencing outflow resistance.15 Research shows actin depolymerization coupled with decreased cell-ECM interactions and myosin II phosphorylation within cells of the trabecular pathway increase aqueous humor outflow, consequently decreasing IOP.16,17 The majority of aqueous flowing across the Schlemm’s canal endothelia, researchers believe, passes through micron-sized pores.7 Schlemm’s canal cells are highly contractile, and increased contraction greatly increases their cell stiffness. Altered cell stiffness modifies pore formation, ultimately affecting downstream egress of AH from the eye.7
Outflow Resistance
Dysfunctional outflow resistance is a concept that is critical to the glaucomatous disease process. A normal reduction of aqueous outflow facility through the TM is approximately 7% to 10% per decade. Primary open angle glaucoma (POAG) patients, however, have increased resistance to outflow.9 Substantial evidence supports a link between cytoskeletal integrity within the cells of the trabecular pathway and aqueous humor outflow through that route.16
 |
| Gonioscopic view of the TM, the initial point of aqueous humor outflow through the conventional outflow pathway and thus a major regulator of IOP. ROCK inhibitors are the first drug class to directly target trabecular outflow, specifically affecting the smooth muscle-like cells of the TM. This novel mechanism results in increased aqueous outflow and subsequent decreased IOP. |
Histological changes in POAG patients that may contribute to decreased aqueous outflow include a declining number of TM cells, increased and changed ECM components, deposition of extracellular plaques and stiffening of the TM with decreased contractility force of the elastic fibers.13,18,19
Specifically, as the smooth muscle–like properties of TM cells likely facilitate dynamic tissue restructuring, the marked loss of these cells in glaucoma leads to fusion and thickening of the trabecular lamellae, which impairs its function.14,19 The deposition of extracellular plaques within glaucomatous JXT ECM is similar to the characteristics of the process of fibrosis. These aberrant accumulations adhere to the sheaths of the elastic fibers and their connections to the inner wall endothelium of Schlemm’s canal.13,19
Researchers believe these increased junctional adherences between the cells of Schlemm’s canal and accumulated ECM underlie the increased resistance to aqueous outflow.20 Bolstering this is the fact that subcortical Schlemm’s canal cell stiffness is elevated, by as much as 50%, in glaucomatous eyes.20 This increased stiffness correlates with decreased pore density, impairing the egress of AH from the eye.20
Current Treatment
The current guidelines for POAG treatment suggest first-line medical therapy with hypotensives.21 Multiple classes of medications are used to lower IOP by affecting both the production and outflow of aqueous humor. These include prostaglandin analogues (PGAs), beta-blockers, carbonic anhydrase inhibitors, alpha adrenergic agonists and miotics.14
PGAs are the most efficacious at lowering IOP, with minimal differences amongst medications in that class.14,22 PGAs work primarily by increasing uveoscleral outflow and, although some studies have shown their ability to alter resistance in trabecular outflow, it is considered minimal.23-26 Uveoscleral outflow, by direct measurement in humans, accounts for approximately 10% to 20% of outflow under normal conditions.4
In the United States, the only currently available medications having a mechanical effect on the conventional outflow pathway are miotics. These agents work by contracting the ciliary muscle, subsequently increasing mechanical pull on the TM, altering the meshwork’s parasellar spaces and leading to an enhanced aqueous outflow.7,16 Due to their significant ocular side effects and frequent dosing schedule, miotics are considered a third-line treatment option.27 Accordingly, today, they are in limited use.7 This underscores a need for drugs that might directly target this pathway with less deleterious side effects.7,28
Trabecular meshwork outflow tissues are avascular and dependent on the AH to supply antioxidants, growth factors and nutrients. Current first-line therapies that suppress aqueous production or enhance uveal/scleral outflow have the potential to decrease the supply of AH nutrients across the outflow tissues.29 In the short-term, reduction of IOP protects the optic nerve; but, in the long-term, nutrient deprivation may induce a greater than normal degradation of the trabecular outflow pathway—with deterioration causing a greater risk of increased IOP over time.29
A Different Approach
The Rho subgroup of the Ras superfamily is made up of multiple small guanosine triphosphate (GTP)-binding proteins (RhoA, RhoB, RhoC).14 Rho GTPase, participates in signaling pathways leading to the formation of actin stress fibers and focal adhesions.30 Rho GTPase is activated in response to growth factors, mechanical stretching, cytokines and extracellular matrix. Rho plays a critical role in a multitude of cellular processes associated with cytoskeletal rearrangements. These include cell morphology, cell motility, cytokinesis, apoptosis and, most notably, smooth muscle contraction.31,32 Multiple Rho target molecules have been identified as downstream Rho effectors, including Rho-associated coil-forming protein serine/threonine kinases labelled as ROCK-1 and its isoform known as ROCK-2.33-35 Stimulating the Rho pathway, specifically the aforementioned ROCK-1 and ROCK-2, enhances phosphorylation of the myosin light chain, thus increasing the contractility of those fibers.16 Upregulation of the Rho pathway has been proposed to play a role in an assortment of diseases, including; asthma, cancer, cardiovascular hypertrophy, diabetes mellitus, erectile dysfunction, hyper-proliferative diseases, hypertension, inflammatory diseases, pulmonary hypertension, renal disease and vasospasm.7
| Rho Kinase Inhibitors Approved or Currently in Active Clinical Trial | |||
| K-115 (Glanatec) | Approved September 2014 (Japan) | D. Western Therapeutics Institute/Kowa Company | ROCK inhibition |
| AR-13324 (Rhopressa) | Phase III | Aerie Pharmaceuticals | ROCK/NET inhibition |
| PG-324 (Roclatan) | Phase III | Aerie Pharmaceuticals | Combination ROCK/NET inhibitor and PGA |
| AMA-0076 | Phase II | Amaken Therapeutics, Belgium | ROCK inhibition |
| Note: A minimum of 8 Rho kinase inhibitor compounds previously in development have been discontinued. This underscores the fact that not all ROCK inhibitors are equally efficacious.14 | |||
This has led to substantial research on the physiological consequences of ROCK inhibition on smooth muscle tone and cytoskeletal stability and its subsequent treatment effects.7
Rho GTPase in Glaucoma
Activation, and sustained activity of, Rho GTPase in the TM cells and other cell types of the AH outflow pathway increases resistance to aqueous humor drainage.15,36 This is associated with increased actin stress fibers and cell adhesive interactions, triggering expression of various ECM proteins and cytokines involved in regulating ECM synthesis.15,36 These changes influence AH drainage working in a feedback response. The discovery of this feedback response uncovered the potential interaction among ECM protein expression, actomyosin contraction and Rho GTPase activity, and their influence on AH drainage through the TM and homeostasis of IOP.36
When glaucomatous optic nerve heads were compared with their non-glaucomatous counterparts, significantly elevated levels of RhoA protein were detected, suggesting RhoA plays a role in the pathophysiology of glaucoma.8 Additionally, endothelin 1 was found in higher concentrations in glaucomatous eyes. Endothelin 1 is a ligand in TM cells that mediates reorganization of the actin cytoskeleton and cell contraction to adjust aqueous outflow. Endothelin 1 is also a major upstream activator of the Rho and Rho-associated protein kinase signaling pathway.14 These findings led researchers to conclude that pharmacological manipulation of the Rho GTPase signaling pathway could prove useful in relaxing the cellular actomysin system, resulting in cell shape alterations and cellular relaxation that results in a downstream increase in intracellular space and a decrease in the resistance to AH outflow.7
ROCK Inhibitors in Glaucoma
Although the probability of glaucoma-related blindness has decreased substantially over the last 45 years, in part due to improvements in medical therapy, no medication with a new mechanism of action has made it to Phase III trials in approximately 20 years.37,38 Ideally, glaucoma medication should focus on three targets: IOP, outflow facility through the pressure-dependent pathway and retinal ganglion cells (RGC).39
Available glaucoma medications focus on aqueous production inhibition and outflow facility improvement through the unconventional pathway.39 New medications with mechanisms of action that act synergistically with existing therapies could interject added benefits.
| ROCK Inhibition and the Endothelium Recent research has concentrated on ROCK inhibition’s use in corneal disease, specifically endothelial dystrophies. To maintain transparency, central endothelial cell (CEC) density must remain above 400-500cells/mm2.1 In pathological situations such as Fuch’s endothelial dystrophy (FD), the rate of cell loss far exceeds that of normal age-related attrition (about 0.6%/year), often terminating with an absolute residual density of 400 cells/mm2.1,2 Pathophysiology CECs have a severely limited ability to proliferate and, as a result of the dystrophy, undergo compensatory migration and enlargement in an attempt to maintain function.3,4 The end result of this endothelial decompensation is often irreversible corneal swelling and loss of transparency with an accompanying reduction in vision.2,5,6 At present, the only curative intervention available is transplantation.2,5 Block the ROCK Endothelial keratoplasty accounted for more than 40% of transplants performed in 2009 and 2010, rising to become the most commonly performed type of keratoplasty in the last three years, approaching 55% in 2014.6,7 Although investigators suggest transplantation of cultivated CECs may be a viable intervention for patients with severe endothelial dystrophies, a less-invasive treatment, such as eye drops, would be more appropriate in early stages of endothelial disease.3 The ROCK inhibitor Y-27632 has shown promise in treating endothelial decompensation, as it works to enhance cell adhesion and accelerate proliferation of the CECs by inhibiting dissociation-induced apoptosis.4,8-12 In a study of four patients scheduled for Descemet stripping automated endothelial keratoplasty, researchers demonstrated the efficacy of Y-27632 eye drops, preceded by trans-corneal freezing, in treating central corneal edema caused by late-onset Fuch’s corneal dystrophy.4,12 Y-27632 proved effective for the recovery of corneal transparency and the gradual reduction of corneal thickness for up to six months after treatment, with one patient’s vision recovering from 20/100 to greater than 20/20.4,12 That patient maintained endothelial function and vision for up to 24 months post treatment.13 A recent preliminary study has also demonstrated that ROCK inhibitor eye drops can treat post cataract surgical corneal edema.5 The investigators concluded the development of such drops for acute corneal damage would be useful in reducing the incidence of bullous keratopathy (PBK) and aphakic bullous keratopathy (ABK).5 1. Joyce, NC. Proliferative capacity of corneal endothelial cells. Exp Eye Res. 2012;95(1):16–23. 2. Bourne W. Biology of the corneal endothelium in health and disease. Eye. 2003;17: 912-8. 3. Okumura N, Koizumi N, Kay E, et al. The ROCK inhibitor eye drop accelerates corneal endothelium wound healing. Invest Ophthalmol Vis Sci. 2013;54:2493-502. 4. Lee J, Kay E. FGF-2-induced wound healing in corneal endothelial cells requires cdc42 activation and rho inactivation through the phosphatidylinositol 3-kinase pathway. Invest Ophthalmol Vis Sci. 2006;47:1376–86. 5. Okumura N, Inoue R, Okazaki Y, et al. Effect of the Rho-kinase inhibitor Y-27632 on corneal endothelial wound healing. Invest Ophthalmol Vis Sci. 2015;56:6067–74. 6. Anshu A, Price M, Tan D, Price F. Endothelial keratoplasty: a revolution in evolution. Surv Ophthalmol. 2012;57:236-52. 7. Eye Bank Association of America. 2014 Eye Banking Statistical Report. Washington, DC; Eye Bank Association of America;2014. 8. Okumura N, Ueno M, Koizumi N, et al. Enhancement on primate corneal endothelial cell survival in vitro by a ROCK inhibitor. Invest Ophthalmol Vis Sci. 2009;50:3680-7. 9. Koizumi N, Okumura N, Ueno M, Knioshita S. New therapeutic modality for corneal endothelial disease using rho-associated kinase inhibitor eye drops. Cornea 2014;33(Suppl):S25–S31. 10. Okumura N, Nakano S, Kay E, et al. Involvement of cyclin D and p27 in cell proliferation mediated by ROCK inhibitors Y-27632 and Y-39983 during corneal endothelium wound healing. Invest Ophthalmol Vis Sci. 2014;55:318–29. 11. Pipparelli A, Arsenijevic Y, Thuret G, et al. ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS One. 2013;8:e62095. 12. Koizumi N, Okumura N, Ueno M, et al. Rho-associated kinase inhibitor eye drop treatment as a possible medical treatment for Fuchs corneal dystrophy. Cornea. 2013;32:1167–70. 13. Ljubimov AV, Saghizadeh M. Progress in corneal wound healing. Prog Retin Eye Res 2015;49:17-45. |
The potential role of Rho GTPases in regulating aqueous humor outflow was first proposed in 2001 when western blot analysis revealed the presence of ROCK in human TM cells and bovine ciliary muscle tissue.30 Now it is understood that the actions of ROCK-1 and ROCK-2 have been shown to alter the cell shape and ECM, increasing the contractility of the TM cells decreasing aqueous humor (AH) outflow facility.6 Accordingly, when ROCK-1 and ROCK-2 are inhibited, research shows IOP is reduced and outflow facility is increased.40 Study authors speculate this effect resulted from ROCK inhibitor-induced retraction of TM cell bodies, disruption of actin bundles and impairment of focal adhesion formation.19 Additional investigations demonstrate that ROCK inhibitors induce changes in cell-cell junction associated proteins and actin cytoskeleton in Schlemm’s canal endothelial cells and cell permeability, leading to improved outflow facility.41 Succinctly, ROCK-based therapies have a mechanism of action that works via relaxation of the contractile tone of the trabecular tissue in the outflow pathway by altering the cell morphology through cytoskeletal disassembly.7
While reducing IOP alone does not seem to halt progressive visual field loss in all patients, specifically those with normal tension glaucoma (NTG) where apoptotic effects may also be in play, reducing damage to the RGCs caused by IOP-independent risk factors might also be useful.42 ROCK inhibitors are known to enhance the survival of RGCs post ischemic injury.43 Researchers investigated the effect of K-115, a novel Rho-kinase inhibitor, on the survival of mice RGC after death was induced by optic nerve crush, finding that survival increased by 34% when K-115 was administered orally.42 Given these results, the authors speculate that suppression of Rho activity has the potential to be a new neuroprotective treatment for glaucoma. ROCK inhibitors may additionally increase blood flow by inhibiting calcium sensitization and relaxing vascular smooth muscles. This effect has been noted in both the conjunctiva and retina and proposed effects directly on the optic disc blood vessels may slow the progression of glaucomatous optic neuropathy.44
According to clinical trial registries in the United States, Europe and Japan, seven different selective rho kinase inhibitors have been tested in human clinical trials.29 ROCK inhibitors have been shown to be effective in lowering the intraocular pressure in human trials, with an IOP reduction ranging from 2.9mm Hg to 6.1mm Hg.29,43,45-48
ROCK Inhibitors In Use
Since Rho-kinase inhibitors have a different mechanism of action than currently available medications, they may work synergistically with the those options. Research demonstrates ROCK inhibitors work successfully in both fixed-dose combination with and additively to travaprost, latanoprost and timolol respectively.29,47 IOP reduction ranged from 9mm Hg to 12mm Hg in a fixed combination of Rho-kinase inhibitor/travoprost, and trials where ROCK inhibitors were added independently to current therapies showed a significantly greater change in mean IOP than when placebo was added.29,47
The most common side effect of ROCK inhibitors is ocular hyperemia, with an incidence of up to 65% in clinical trials.29 Hyperemia is due to relaxation of the smooth muscle cells of the conjunctival blood vessels and is transient, resolving in a few hours.43-46,48 Once-daily dosing at night minimizes this, reducing the incidence of hyperemia to 11% as the hyperemia resolves overnight.29
Additional mild to moderate adverse events reported with ROCK inhibitors included: punctate keratitis, photophobia, headache, abdominal pain, mild hepatic dysfunction, asthma and nasopharyngitis.46,47 Notably, no severe adverse events have been reported in clinical trials. Glanatec (ripasudil, Kowa), K-115, approved for glaucoma and ocular hypertension in September 2014 in Japan, has been shown to induce guttae-like findings.49 These are due to the formation of protrusions along the endothlium cell-cell borders. They appear to be transient. Corneal endothelial function markers, corneal thickness and corneal volume have been reportedly unaffected, and no endothelial cell death has been recorded.49
Investigators hypothesize that ROCK inhibitors also lower IOP by reducing episcleral venous pressure (EVP). A recent preclinical study has shown that AR-13324, the first of a new class of ocular hypotensive compounds that inhibits both Rho kinase and the norepinephrine transporter, (ROCK-NET) produces a statistically significant lowering of EVP in rabbits.50 The authors speculate that a similar effect in humans would offer an additional new mechanism of action for patients with glaucoma.
A Phase II study comparing AR-13324 with latanoprost showed AR-13324 to be less effective by approximately 1mm Hg, reducing the IOP by a substantial 5.7mm Hg on average.51 ROCK-NETs work by inhibiting both Rho kinase and norepinephrine transporter to increase outflow and by decreasing the production of AH.52 The advancement of this medication to Phase III trials is highly promising and will hopefully yield a novel treatment for patients in the United States.
The potential impact of ROCK-inhibition in the management of glaucoma and FD appears to be promising. These medications, with their novel mechanism of action, will add to the therapeutic collection with the promise of improving vision and quality of life for our patients.
Dr. Rixon is an attending optometrist at the Memphis VA Medical Center in Tennessee.
Dr. Gurwood is a professor at Salus University in Elkins Park, Pa.
|
1. Weinreb R, Aung T, Medeiros F. The pathophysiology and treatment of glaucoma: a review. JAMA. 2014 May;311(18):1901-11. 2. Janssen S, Gorgels T, Ramdas W, et al. The vast complexity of primary open angle glaucoma: disease genes, risks, molecular mechanisms and pathobiology. Prog Retin Eye Res. 2013;37:31-67. 3. Wilson M, Singh K. Intraocular pressure: does it measure up? The Open Ophthalmology Journal. 2009;3:32-7. 4. Alm A, Nilsson S. Uveoscleral outflow: a review. Experimental Eye Research. 2009;88:760-8. 5. Brubaker, R. Targeting outflow facility in glaucoma management. Surv Ophthalmol. 2003;48:Suppl 1: S17-20. 6. Stamer W, Braakman S, Zhou E, et al. Biomechanics of Schlemm’s canal endothelium and intraocular pressure reduction. Prog in Retinal Eye Research. 2015;44:86-98. 7. Challa P, Arnold J. Rho-kinase inhibitors offer a new approach in the treatment of glaucoma. Expert Opin Investig Drugs. 2014;23(1):81-95. 8. Goldhagen B, Prola A, Epstein D, Rao P. Elevated levels of RhoA in the optic nerve head of human eyes with Glaucoma. J Glaucoma. 2012;21:530-8. 9. Johnson M. What controls aqueous humor outflow resistance? Exp Eye Res. 2006;82(4):545-57. 10. Tamm E. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res. 2009;88:648-55. 11. Lutjen-Drecoll E. Functional morphology of the trabecular meshwork in primate. Prog Retin Eye Res. 1999;18:91-119. 12. Tian B, Gabelt B, Geiger B, Kaufman P. The role of the actomyosin system in regulating trabecular fluid outflow. Exp Eye Res. 2009;88:713-7. 13. Keller K, Acott T. The juxtacanalicular region of ocular trabecular meshwork: A tissue with a unique extracellular matrix and specialized function. J Ocul Biol. 2013 June;1(1):3. 14. Wang S, Chang R. An emerging treatment option for glaucoma: rho kinase inhibitors. Clin Ophthalmol. 2014:8;883–90. 15. Pattabiraman P, Maddala R, Rao P. Regulation of plasticity and fibrogenic activity of trabecular meshwork cells by rho GTPase signaling. J Cell Physiol. 2014 July;229(7):927-42. 16. Pattabiraman P, Rao P. Mechanistic basis of rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am J Physiol Cell Physiol. 2010;298:C749-763. 17. Kaufman P. Enhancing trabecular outflow by disrupting the actin cytoskeleton, increasing uveoscleral outflow with prostaglandins and understanding the pathophysiology of presbyopia: Interrogating Mother Nature: asking why, asking how, recognizing the signs, following the trail. Eye Exp Res. 2008;86(1):3-17. 18. Keller K, Aga M, Bradley J, et al. Extracellular matrix turnover and outflow resistance. Exp Eye Res. 2009;88(4):676-82. 19. Tektas O, Lutjen-Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. 2009;88(4):769-75. 20. Overby D, Zhou E, Vargas-Pinto R, et al. Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc Natl Acad Sci USA. 2014;111(38):13876-81. 21. Li T, Lindsley K, Rouse B, et al. Comparitive effectiveness of first-line medications for primary open-angle glaucoma: A systematic review and network meta-analysis. Ophthalmology. 2016 Jan;123(1):129-40. 22. Parrish R, Palmberg P, Sheu W, XLT Study Group. A comparison of latanoprost, bimatoprost, and travoprost in patients with elevated intraocular pressure: a 12-week, randomized, masked-evaluator multicenter study. Am J Ophthalmol. 2003;135(5):688-703. 23. Alm A, Villumsen J. PhXA34, a new potent ocular hypotensive drug. A study on dose-response relationship and on aqueous humor dynamics in healthy volunteers. Arch Ophthlmol. 1991;109(11):1564-8. 24. Toris C, Camras C, Yablonski M. Effects of PhXA41, a new prostaglandin F2, analog on aqueous humour dynamics on human eyes. Ophthalmology. 1993;100:1297-304. 25. Toris C, Gabelt B, Kaufman P. Update on the mechanism of action of topical prostaglandins for intraocular pressure reduction. Surv Ophthalmol. 2008;53(S1):S107-120. 26. Fung D, Whitson J. An evidence-based review of unoprostene isopropyl ophthalmic solution 0.15% for glaucoma: place in therapy. Clin Ophthalmol. 2014;8:543-54. 27. Lee D, Higginbotham E. Glaucoma and its treatment: a review. Am J Health-Syst Pharm. 2005;62:691-9. 28. Tanihara H, Inatani M, Honjo M, et al. Intraocular pressure-lowering effects and safety of topical administration of a selective rock inhibitor, SNJ-1656, in healthy volunteers. Arch Ophthalmol. 2008;126(3):309-15. 29. Kopczynski C, Epstein D. Emerging trabecular outflow drugs. J Ocul Pharmacol Ther. 2014;30(2-3):85-7. 30. Honjo M, Tanihara H, Inatani M, et al. Effects of Rho-associated protein kinase inhibitor Y-27632 on intraocular pressure and outflow facility. Invest Ophthalmol Vis Sci. 2001;42:137-44. 31. Honjo M, Inatani M, Kido N, et al. Effects of protein kinase inhibitor, HA1077, on intraocular pressure and outflow facility in rabbit eyes. Arch Ophthalmol. 2001;119:1171-8. 32. Okumura N, Ueno M, Koizumi N, et al. Enhancement on primate corneal endothelial cell survival in vitro by a ROCK inhibitor. Invest Ophthalmol Vis Sci. 2009;50:3680-7. 33. Ishizaki T, Naito M, Fujisawa K,et al. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS letter. 1997;404:118-24. 34. Leung T, Chen X, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313-27. 35. Amano M, Chihara K, Kimura K, et al. Formation of actin stress fibers and focal adhesion enhanced by rho-kinase Science. 1997;275(5304):1308-11. 36. Zhang M, Maddala R, Rao P. Novel molecular insights into RhoA GTPase-induced resistance to aqueous humor outflow through the trabecular meshwork. Am J Physiol Cell Physiol. 2008;295(5):C1057-70. 37. Malihi M, Moura Filho E, Hodge D, Sit A. Long-term trends in glaucoma-related blindness in Olmsted County, Minnesota. Ophthalmology. 2014;21(1):134-41. 38. Alm A, Camras C, Watson P. Phase III latanoprost studies in Scandinavia, the United Kingdom and the United States. Surv Ophthalmol. 1997;41 Suppl 2:S105-10. 39. Brubaker R. Introduction: Three Targets for Glaucoma Management. Surv Ophthalmol. 2003;48 Suppl 1:S1-2. 40. Tham YC, Li X, Womg TY, et al. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology. 2014;121:2081-90. 41. Kameda T, Inoue T, Inatani M, et al. The effect of rho associated protein kinase inhibitor on monkey Schlemm’s canal endothelial cell Invest. Ophthalmol Vis Sci. 2012 53(6):3092-103. 42. Yamamoto K, Maruyama K, Himori N, et al. The novel Rho Kinase (ROCK) inhibitor K-115: A new candidate drug for neuroprotective treatment in glaucoma. Invest Ophthalmol Vis Sci. 2014:55(11):7126-36. 43. Williams R, Novack G, van Haarlem T, et al. Ocular hypotensive effect of the rho kinase inhibitor AR-12286 in patients with glaucoma and ocular hypertension. Am J Ophthalmol. 2011;152(5):834-4. 44. Inoue T, Tanihara H. Rho-associated kinase inhibitors: A novel glaucoma therapy. Prog Retin Eye Res. 2013;37:1-12. 45. Tanihara H, Inoue T, Yamamoto T, et al. Additive intraocular pressure-lowering effects of the rho kinase inhibitor ripasudil (K-115) combined with timolol or latanoprost A report of two randomized clinical trials. JAMA Ophthalmol. 2015;133(7):755-61. 46. Tanihara H, Inoue T, Yamamoto T, et al. Phase 1 clinical trials of a selective rho kinase inhibitor, K-115. JAMA Ophthalmol. 2013;131(10):1288-95. 47. Garnock-Jones K. Ripasudil: First global approval. Drugs. 2014;74:2211-5. 48. Inoue T, Tanihara T, Tokushige H, Arale M. Efficacy and safety of SNJ-1656 in primary open-angle glaucoma or ocular hypertension. Acta Ophthalmol. 2015;93(5):e393-5. 49. Okumura N, Okazaki Y, Inoue R, et al. Rho-associated kinase inhibitor drop (ripasudil) transiently alters the morphology of corneal endothelial cells. Invest Ophthalmol Vis Sci. 2015;56(12) 7560-7. 50. Kiel J, Kopczynski C. Effect of AR-13324 on episcleral venous pressure in dutch belted rabbits. J Ocul Pharmacol Ther. 2015;31(3):146-51. 51. Bacharach J, Dubiner H, Levy B, et al. Double-masked, randomized, dose-response study of AR-13324 versus latanoprost in patients with elevated intraocular pressure. Ophthalmology. 2015;122:302-7. 52. Wang R, Williamson J, Kopcyznski C, Serle J. Effect of 0.04% AR-13324, a ROCK and norepinephrine transporter inhibitor, on aqueous humor dynamics in normotensive monkey eyes. J Glaucoma. 2015;24:51–4. |
